
Abstract
Keywords
Introduction
Aneurysmal subarachnoid hemorrhage (aSAH) accounts for only 5-10% of all strokes, 1 but the associated morbidity and socioeconomic burden exceed those of ischemic strokes due to the younger age of affected patients. 2 In addition to early brain injury within the first 72 hours, 3 patient outcomes are determined by delayed secondary brain injury (SAH-SBI), which often occurs between days 3 and 14 after aneurysm rupture. 4 Two-thirds of patients develop angiographic vasospasm (aVSP) in the large cerebral arteries. 5 Delayed cerebral ischemia (DCI) with radiologic demarcation of ischemic brain areas and clinically evident delayed ischemic neurologic deficits (DINDs) are found in one-third of patients with aSAH. 6 The pathophysiology of SAH-SBI is multifactorial, involving macro- and microvascular dysfunction, microthombosis, neuroinflammation, neuronal apoptosis, and pathological electrical brain activity.4,7–10 The delay between aneurysm rupture and the onset of SAH-SBI provides a window of opportunity for preventive and therapeutic interventions. However, to date, the only preventive intervention for SAH-SBI that has been shown to moderately improve neurological outcomes is oral nimodipine.11,12 In already symptomatic patients, the therapeutic options are limited to noncausal rescue therapies to improve cerebral perfusion, such as the adaptation of blood pressure targets or angioplasty.11,12 Therefore, methods to identify patients at high risk for SAH-SBI as well as novel therapeutic targets represent an unmet need.
Hemoglobin has been implicated as a potential driver of SAH-SBI for decades.7,13,14 However, only recent preclinical studies have begun to demonstrate clear pathophysiological links between Hb and mechanisms of SAH-SBI.15,16 We propose the following pathophysiological sequence to underlie the increased risk for SAH-SBI: red blood cells (RBCs) decompose in the subarachnoid hematoma, cell-free Hb is generated in the CSF (CSF-Hb), Hb tetramers dissociate into dimers, and finally small CSF-Hb dimers delocalize into vulnerable anatomical sites of the cerebral arteries and brain. There, nitric oxide (NO) scavenging promotes vasoconstriction15,17 and potentially oxidative neuronal damage. In experimental models, these adverse effects of CSF-Hb were mitigated by intraventricular administration of the Hb scavenger protein haptoglobin. 15 Collectively, this prior evidence suggests CSF-Hb as a biomarker that may allow the risk of SAH-SBI to be estimated by directly monitoring a potentially targetable pathophysiological process.
The main objective of this study was to investigate the clinical association of CSF-Hb with SAH-SBI and the diagnostic accuracy of daily CSF-Hb measurements. Furthermore, we performed an in-depth CSF proteome study and ex-vivo functional assays to rationalize the role of CSF-Hb as a pathophysiological driver of and potential therapeutic target for SAH-SBI.
Material and methods
General
This study was approved by and all procedures were performed in accordance with the Ethics Committee of the Canton of Zurich, Switzerland (KEK ZH 2016-00439) and with the guiding principles of the 2008 Declaration of Helsinki. Written informed consent was obtained from all patients or their legal representatives. The results are reported in accordance with the STROBE statement (von Elm et al., 2007).
Study population
Clinical data and CSF samples were obtained from a prospective consecutive cohort of aSAH patients admitted to the Neurointensive Care Unit of the Department of Neurosurgery, University Hospital Zurich over a 3-year period (April 2017 to March 2020). The eligibility criteria are given in the Supplementary Methods.
Clinical data acquisition
Patient data collection was performed by the treating physician blinded to the CSF measurements. Baseline features of the patients were assessed at the time of diagnosis and are given in the Supplementary Methods (Supplementary Figure 1(a)). Clinical data was collected prospectively during the 14-day high-risk phase for SAH-SBI based on standardized monitoring at the Neurointensive Care Unit and as part of the 3-month follow-up. General patient management conformed to current guidelines of the Neurocritical Care Society and the American Heart Association.11,12 The presence of DIND, DCI and aVSP was assessed on a daily basis during the 14-day sampling period. The definitions of DIND, DCI and aVSP are given in the Supplementary Methods. SAH-SBI was defined as the composite outcome of DIND, DCI and aVSP. In addition, flow velocity in the bilateral middle, anterior and posterior cerebral arteries was assessed daily using Transcranial Doppler (TCD) sonography. Complications were assessed daily during the 14-day sampling period (details see Supplementary Methods). Chronic hydrocephalus (ventriculoperitoneal shunt dependency) and functional status (Glasgow Outcome Scale-Extended [GOSE] and modified Rankin Scale [mRS]) were evaluated at the 3-months follow-up.
Subarachnoid hematoma segmentation and volumetric analysis
Based on the initial CT scan, the volume of the subarachnoid hematoma was manually delineated using 3DSlicer 4.11.0. 18 The preprocessing prior to delineation consisted of brain extraction whereby the largest cavity from a 200 HU thresholded skull mask was extracted. 19 Subsequently, the brain mask was multiplied with the original CT scan and median filtered with a 5×5×3 kernel (Supplementary Figure 1(b)). The volume of the hemorrhage was calculated by multiplying the voxel size by the number of segmented voxels.
CSF sampling
CSF samples were collected from the EVD by investigators blinded to the clinical data. The decision regarding the insertion of an EVD was made independently of this study and according to the clinical standard of care. 1 ml of CSF was sampled on a daily basis at approximately the same time (morning). A sampling duration from day 1 (the day after aSAH) to day 14 was used. If the patient's EVD was removed prior to day 14, the CSF sampling was discontinued. Immediately after sample collection, the CSF was centrifuged (Capricorn CEP 2000 Benchtop centrifuge, Capricorn labs, UK) at 1500 ×g for 15 minutes, and the supernatant was stored at -80°C for further analysis (Supplementary Figure 1(c)).
Spectrophotometry of CSF-Hb and heme metabolites
Absorption spectra in the visual range between 350 and 650 nm of all CSF samples were measured on a Shimadzu UV-1800 spectrophotometer (Shimadzu, Japan). Quantification of different Hb species and their metabolites in the CSF (oxyHb [hereafter referred to as CSF-Hb], metHb, bilirubin, biliverdin) was performed using spectral deconvolution. Therefore, extinction curves for the individual substances with known concentrations were fitted to the extinction curve of CSF using a nonnegative least squares algorithm as described previously. 20
LC-MS/MS CSF proteomic analysis
Using LC-MS/MS based label-free quantification, the CSF proteomes of 18 patients were obtained at five sequential time points during the observation period (0, 0.5, 1, 1.5 and 2 weeks after aSAH) (Supplementary Figure 1(c)). Details of the proteomics workflow, sample preparation and data analysis are included in the Supplementary Methods. Temporal changes in the CSF proteome were clustered using k-means analysis, whereby the optimal number of clusters was determined visually with an elbow diagram (Supplementary Figure 1(d)) using the factoextra package. 21
Neurovascular function
The neurovascular function assay (Supplementary Figure 1(e)) was performed using fresh porcine basilar arteries obtained from a local abattoir (n = 12). 15 Methodological details are given in the Supplementary Methods and the sequence of the experiment is illustrated in Supplementary Figure 1(f). An initial series of experiments was conducted to evaluate the vasoconstrictive potential of patient CSF during the high-risk phase for SAH-SBI and to assess the specific role of Hb in this process. For this purpose, patient CSF collected between days 3 and 14 after aSAH was selectively depleted of CSF-Hb using a haptoglobin affinity column as previously described. 15 In the initial phase of the experiment, the vessels were immersed in Hb-depleted CSF. Subsequently, the precise amount of Hb that had been removed from the CSF was restored, in order to determine the specific impact of CSF-Hb on vascular tension. In a second series of experiments, the influence of increasing Hb exposure on vascular tension was assessed. For that purpose, the vessels were immersed in Krebs-Henseleit buffer and exposed to increasing concentrations of Hb (10−4 M to 10−8 M Hb in half log10 steps). To evaluate the effect of the hemoglobin-scavenger haptoglobin, a third series of experiments was performed with identical Hb concentrations, but an equimolar amount of haptoglobin was added.
Hb, haptoglobin, hemopexin and reconstituted lipoprotein (rLP)
Hb for use in ex-vivo experiments was purified from expired human blood concentrates as previously described. 22 Hb concentrations were determined by spectral deconvolution as described above and are given as molar concentrations of total heme (1 M Hb tetramer is equivalent to 4 M heme). For all Hb used in these studies, the fraction of ferrous Hb (HbFe2+O2) was always greater than 98%, as determined by spectrophotometry. Purified haptoglobin (phenotype 1-1) and hemopexin from human plasma, as well as reconstituted lipoprotein (rLP) were obtained from CSL Behring, Bern, Switzerland.
Thiobarbituric acid-reactive substances (TBARS) assay
The oxidative potential of the CSF samples was quantified by measuring the formation of malondialdehyde (MDA), the final product of lipid peroxidation, after incubation with rLP (Supplementary Figure 1(g)). Methodological details are given in the Supplementary Methods.
Statistical analyses
Statistical analyses were performed using R 3.6.3. 23 The detailed statistical methods as well as the versions of the used R-packages are given in Supplementary Methods. Descriptive statistics are presented as absolute numbers (n) and proportions (%) for categorical variables, whereas continuous variables are shown as mean and standard deviation (SD). The Shapiro-Wilk test was used to test for normality. Due to the exploratory nature of the study, no level of statistical significance was defined; instead, the results were interpreted based on the level of evidence for an association as follows: p < 0.001: very strong evidence; p < 0.01: strong evidence; p < 0.05 evidence; p < 0.1 weak evidence; and p > 0.1: no evidence. 24
Data and code availability
The authors confirm that the data supporting the findings of this study are available within the article and its Supplementary Materials. In addition, all anonymized datasets used in this study along with the statistical code are provided in the Supplementary Materials (for an overview, see Supplementary Table 3). The use of dynamic reporting guarantees full reproducibility of the results given data and code.
Results
Study population
Out of 52 consecutively screened patients fulfilling the eligibility criteria, 47 were included, since for five patients informed consent was refused. Table 1 summarizes the baseline features of the patient cohort.
Baseline cohort characteristics. Demographic, clinical and radiological cohort features at baseline are shown.

aMaximal diameter in mm.
bVolume in cm3.
BNI: Barrow Neurological Institute; GCS: Glasgow Coma Scale; SD: standard deviation; WFNS: World Federation of Neurosurgical Societies.
Spectrophotometry demonstrates delayed accumulation of CSF-Hb and heme metabolites in patient CSF
We used spectrophotometry with spectral deconvolution to quantify CSF-Hb, its downstream metabolites biliverdin and bilirubin, and the primary Hb oxidation product metHb (Figure 1(a)). The individual temporal profiles and peak concentrations of CSF-Hb (0.6 µM to 242.2 µM) were highly variable (Supplementary Figure 2). In most patients, CSF-Hb remained very low over the first 2 to 3 days after aSAH, followed by a strong increase, a plateau between days 9 and 12, and a decrease thereafter. Bilirubin was already elevated on day 1 and increased at the greatest rate within the first 3 to 5 days before it reached a plateau. Levels of the intermediate metabolite biliverdin markedly increased from day 4 and peaked on day 12. MetHb levels showed a delayed increase with a peak on day 11 after aSAH. Hence, high levels of CSF-Hb develop with a delay after the acute bleeding and coincide with the high-risk phase for SAH-SBI.

Changes in the cerebrospinal fluid proteome after aneurysmal subarachnoid hemorrhage. (a) Temporal cerebrospinal fluid (CSF) oxyhemoglobin (CSF-Hb), bilirubin, biliverdin and methemoglobin (metHb) profiles. (b) K-means clustering of proteins in the CSF after aneurysmal subarachnoid hemorrhage (aSAH) identified with LC-MS/MS. (c) Volcano plot showing the overall fold change and combined p-value for the CSF proteome. The color indicates the respective cluster, the size of each dot represents the raw mean intensity of the protein (not normalized). (d) Enrichment plot showing the top negatively enriched hallmark gene sets for coagulation identified by gene set enrichment analysis (GSEA) of the CSF proteome after aSAH. (e) Temporal course of the normalized protein intensities for haptoglobin (HP), haptoglobin-related protein (HPR), hemopexin (HPX) and albumin (ALB) in the CSF after aSAH. (f) Enrichment plot of the top positively enriched hallmark gene set for glycolysis identified by GSEA of the CSF proteome after aSAH. (g) Temporal course of the normalized protein intensities for carbonic anhydrase 1 (CA1), carbonic anhydrase 2 (CA2), catalase (CAT) and aldolase A (ALDOA) in the CSF. (h) Enrichment plot of the hallmark gene set for heme metabolism identified by GSEA of the CSF proteome after aSAH. (i) Temporal course of the normalized protein intensities for CD163, colony stimulating factor 1 receptor (CSF1R), CD14 and metalloproteinase inhibitor 1 (TIMP1) in the CSF. The box in the boxplots bounds the interquartile range (IQR) divided by the median, while the whiskers extend to the highest and lowest value within the 1.5 × IQR, respectively.
LC-MS/MS analysis of CSF proteins delineates erythrolysis and a dynamic macrophage response in the subarachnoid space
We performed a quantitative LC-MS/MS analysis of 85 CSF samples from 18 patients collected at 5 time points after aSAH. Figure 1(b) illustrates the temporal course of the levels of 757 CSF proteins that were assigned to four clusters by a k-means algorithm. Figure 1(c) shows a volcano plot of the normalized signal intensities summed across all samples per patient, providing an overall view of protein accumulation or depletion within the two-week period after aSAH. Cluster 2 (green) comprised proteins whose intensity remained mostly unchanged over time. Cluster 3 (yellow) represents proteins with decreasing intensity over time. These were mainly plasma proteins that likely entered the subarachnoid space with the bleeding and were subsequently cleared from the CSF. GSEA of the ranked proteins assigned the top negatively enriched hallmark gene set to coagulation (ES = -0.66, FDR = 0.007, Figure 1(d)). Figure 1(e) displays the levels of four selected plasma proteins (HP, HPR, HPX and ALB) over time. Cluster 1 (violet) contained proteins that showed pronounced accumulation over time, and cluster 4 (blue) contained proteins whose levels showed a moderate increase. GSEA identified glycolysis (ES = 0.78, FDR = 0.021, Figure 1(f)) and heme metabolism (ES = 0.74, FDR = 0.096, Figure 1(h)) as the top enriched hallmark gene sets, indicating the lysis of RBCs with subsequent release of cytoplasmic proteins and the breakdown of Hb by macrophages, respectively. Figure 1(g) displays levels of the RBC proteins carbonic anhydrase 1 (CA1) and 2 (CA2), catalase (CAT), and aldolase A (ALDOA) over time. Figure 1(i) shows the macrophage proteins CD163, CSF1R, CD14, and TIMP1. The accumulation of these proteins suggests a pronounced macrophage influx into the subarachnoid space within the first days after hemorrhage. Collectively, the proteome dynamics in patient CSF after aSAH reinforced the notion that erythrolysis and an adaptive macrophage response are the two dominant processes in the CSF space after aSAH.
Hematoma volume, day after aSAH and presence of IVH as relevant determinants of CSF-Hb
To estimate determinants of CSF-Hb concentration, we applied a GAM with non-linear spline-fit (4 knots) for time (day after aSAH) and a random-effect for the individual patients. The partial dependence plots are shown in Figure 2(a). Larger aneurysm size was not associated with higher CSF-Hb. Among aneurysms at different locations, middle cerebral artery aneurysms showed strong evidence for a negative association with CSF-Hb (partial dependence = -0.57, SD = 0.20, p = 0.0044). There was very strong evidence for a positive association between hematoma volume and CSF-Hb (p < 0.001). A rendered example of the subarachnoid blood clot is given in Figure 2(b). Volumetric analysis was performed in only 46 of the 47 patients, as one patient had an initial MRI (and not a CT scan) and was therefore excluded from this analysis. There was very strong evidence (p < 0.001) for an association between the time point of sampling (day) and CSF-Hb, whereas the partial dependence was negative in the first 3-5 days after aSAH, and positive in subsequent days. This association most likely reflects the delayed onset of erythrolysis and CSF-Hb liberation in the subarachnoid space after aSAH. Also, there was very strong evidence for a positive association between the presence of IVH and CSF-Hb (partial dependence = 1.1, SD = 0.15, p < 0.001). With IVH, CSF-Hb levels increased early and then reached a plateau, whereas in the absence of IVH, this increase was more delayed and gradual, but a plateau was reached at comparable peak concentrations around day 12 (Figure 2(c)). This most likely represents a CSF compartment effect, since the EVD used to sample the CSF is in direct proximity to the blood clot when IVH is present, while the EVD lies distant to the blood clot, if IVH is absent (illustrated in Figure 2(c)). In summary, these findings indicate that hematoma volume, day after aSAH and the presence of IVH are relevant determinants of CSF-Hb levels.

Determinants of cerebrospinal fluid hemoglobin. (a) Partial dependence of cerebrospinal fluid hemoglobin (CSF-Hb) levels on aneurysm size, aneurysm location, hematoma volume, the presence of intraventricular hemorrhage (IVH), and number of days post-SAH according to a generalized additive model (GAM). (b) Example of 3 D-rendered subarachnoid hematoma. (c)
Increased CSF-Hb levels are associated with SAH-SBI
The cohort outcome parameters representative of SAH-SBI, rescue therapies, complications, mortality, and functional status at the 3-month follow-up are summarized in Supplementary Table 2. The CSF-Hb levels stratified by aVSP, DCI, DIND and SAH-SBI are shown in Figure 3(a) and Supplementary Figure 3. As expected, given their strict clinical definitions, the status of DIND, DCI and aVSP is missing on many time points (denoted as not available (NA)). Supplemental Figure 3 shows the CSF-Hb values of the NAs for DINDs, DCI and aVSPs compared to the CSF-Hb. Within the NAs, the CSF-Hb values were found distributed along the entire measurement spectrum without an unidirectional trend, as opposed to those with or without DIND, DCI or aVSP. A GAM with spline fit (5 knots) for time (day after aSAH) and a random-effect for the individual patients provided very strong evidence (p < 0.001) for a positive association between CSF-Hb and SAH-SBI. The partial dependence of SAH-SBI on CSF-Hb and the day after aSAH are given in Figure 3(b).

Association between cerebrospinal fluid hemoglobin and secondary brain injury after aneurysmal subarachnoid hemorrhage. (a) Cerebrospinal fluid hemoglobin (CSF-Hb) in patients after aneurysmal subarachnoid hemorrhage (aSAH) stratified by angiographic vasospasm (aVSP), delayed cerebral ischemia (DCI), delayed ischemic neurological deficit (DIND) and the composite outcome SAH-related secondary brain injury (SAH-SBI) per day after aSAH (day post-SAH). (b) Partial dependence of SAH-SBI on CSF-Hb (log-scale) and the number of days post-SAH according to a generalized additive model (GAM). (c) Receiver operating characteristic (ROC) curves and area under the curves (AUC) of CSF-Hb for DIND, DCI and aVSP (left). ROC curve of CSF-Hb for SAH-SBI, with the corresponding CSF-Hb measurements. (d) The temporal course of CSF-Hb stratified by GOSE score at the 3-month follow-up. The box in the boxplots bounds the interquartile range (IQR) divided by the median, while the whiskers extend to the highest and lowest value within the 1.5 × IQR, respectively.
To estimate the diagnostic accuracy of daily CSF-Hb measurements to monitor for SAH-SBI, we calculated the ROC curves and AUCs for SAH-SBI and the individual outcomes aVSP, DCI and DIND (Figure 3(c), Supplementary Figure 4(a) to (c)). The high diagnostic accuracy of CSF-Hb remained unaltered in a recalculated model with data limited to the high-risk period (days 4-14), avoiding potential bias introduced by the generally low Hb levels in the first 3 days after aSAH (Supplementary Figure 4(d)). Bilirubin, biliverdin and metHb had a lower diagnostic accuracy than CSF-Hb (Supplementary Figure 4(e) to (h)). Computation of the optimal Youden index yielded a CSF-Hb value of 7.1 µM for SAH-SBI (Figure 3(c)). For the individual outcomes of aVSPs, DCI and DINDs, the optimal Youden indices were 3.4, 5.5, and 7.1 µM, respectively (Supplementary Figure 4(a) to (c)). TCD measurements demonstrated a high specificity for SAH-SBI (0.97 [0.88 to 1.00]), DIND (0.96 [0.87 to 1.00]), DCI (0.93 [0.76 to 0.99]) and aVSP (0.96 [0.78 to 1.00]). However, the sensitivity of daily TCD measurements was minimal (SAH-SBI: 0.28 [0.17 to 0.42], DIND: 0.32 [0.18 to 0.48], DCI: 0.15 [0.03 to 0.38], aVSP: 0.17 [0.05 to 0.37]).
CSF-Hb values stratified by good and poor functional outcome at 3-month follow up based on GOSE and mRS score are shown in Figure 3(d) and Supplementary Figure 4(h), respectively. A GAM with spline fit (4 knots) for time (day after aSAH) and a random-effect for the individual patients provided very strong evidence for a positive association of CSF-Hb to a poor functional outcome at 3-month follow up, as expressed by the GOSE (partial dependence = 0.71, SD = 0.10, p < 0.001) and mRS (partial dependence = 0.72, SD = 0.10, p < 0.001).
Basilar artery vasoconstriction and lipid peroxidation occur in the concentration range of patient CSF-Hb
Our clinical data suggested CSF-Hb as an upstream mediator of SAH-SBI. To further investigate a potential pathophysiological association, we studied whether ex-vivo vasoconstriction and lipid peroxidation with Hb exposure correlated with the range of CSF-Hb concentrations observed in our cohort. We established a new model of vascular function with porcine basilar arteries that were precontracted with PGF2α and dilated by an intrinsic endothelial NO synthase (eNOS) response. This setup allowed us to study the effect of Hb on endogenous NO reserve capacity. In contrast to our prior proof-of-concept studies, 15 which were not designed to validate the pathophysiological effects of predefined Hb concentrations, no supplemental NO donor was used. Figure 4(a) demonstrates the sigmoid dose-response curve, which shows a steep increase in vascular tension between 10−6.5 and 10−6 M Hb and maximum contraction near 10−5 M Hb.

The vasoconstrictive and oxidative potential of hemoglobin. (a) Relative tension dose-response curve for hemoglobin (Hb). (b) Patient Transcranial Doppler (TCD) velocity (max-TCD-scaled) correlated to CSF-Hb levels. The presence of angiographic vasospasms (aVSP) is displayed as a color overlay. (c) Formation of malondialdehyde (MDA) in response to different concentrations of Hb determined by a TBARS assay. (d) Correlation between measured CSF-Hb and the presence of secondary brain injury (SAH-SBI). The box in the boxplots bounds the interquartile range (IQR) divided by the median, while the whiskers extend to the highest and lowest value within the 1.5 × IQR, respectively.
An almost identical curve was obtained when we plotted the max-TCD-scaled values of our patient cohort against the measured CSF-Hb concentrations (Figure 4(b)). The samples from those patients with aVSPs (red dots) in the majority contained CSF-Hb at concentrations of 10−5 M or more.
To investigate the oxidative potential associated with CSF-Hb, we measured the generation of MDA in a mixture of Hb and rLP containing unsaturated phosphatidylcholine, which is the main physiological lipid substrate for in-vivo Hb peroxidation reactions. 20 The dose-response curves shown in Figure 4(c) demonstrates that the steepest increase in MDA formation also occurred with Hb at concentrations between 10−5.5 and 10−5 M with reaction incubation times ranging from 0.5 to 4 h.
Figure 4(d) plots the patient’s CSF-Hb concentrations stratified by the presence of SAH-SBI. This demonstrates that the optimal Youden index of CSF-Hb for SAH-SBI, which arithmetically yields the optimal ratio between sensitivity and specificity, nearly coincides with the CSF-Hb concentration at maximal basilar artery vasoconstriction, the maximum TCD values and the concentration at which the rate of TBARS generation is highest.
Collectively, the results of the vascular function and lipid peroxidation studies reveal critical inflection points for toxic Hb-effects at concentrations overlapping the CSF-Hb concentrations found in patients with SAH-SBI.
Depletion and neutralization of CSF-Hb mitigates pathological vasoconstriction and lipid peroxidation
To mechanistically link CSF-Hb in patients with SAH-SBI, we performed Hb depletion and neutralization experiments. Porcine basilar artery segments that were immersed in CSF depleted of Hb with a haptoglobin affinity column (Figure 5(a)) had a lower relative tension than segments immersed in the same CSF sample in which the original CSF-Hb concentration had been restored with highly purified Hb (Figure 5(b)). This experimental setup controlled for potential nonspecific removal of vasoactive substances by the haptoglobin affinity column. Additionally, we repeated a Hb dose-response experiment to confirm the anti-vasospastic effect of soluble haptoglobin (phenotype 1-1, Hp 1-1). Across the Hb concentration range, Hp 1-1 attenuated the contractile force of basilar arteries (Figure 5(c)). We also quantified the oxidative potential of CSF samples from patients after aSAH. We found variable increases in MDA after lipoprotein exposure between CSF samples that were collected during the high-risk phase (weeks 0.5-2) compared to baseline CSF samples (collected on day 1) (Figure 5(d)). The addition of the Hb-scavenger Hp 1-1 at equimolar concentrations to CSF-Hb effectively attenuated MDA formation. The addition of the heme-scavenger hemopexin at an equimolar concentration to CSF-Hb attenuated MDA formation to an even greater extent. These ex-vivo experiments show the effectiveness of Hb and heme scavengers in mitigating the vasoconstrictive, as well as the oxidative effects of CSF-Hb, and thereby support further evaluation for their clinical application of patients after aSAH.

Protective effects of scavenger proteins against the vascular and oxidative effects of hemoglobin. (a) Depletion of Hb from CSF using a haptoglobin (Hp) affinity column. (b) Vascular tension of vessels immersed in Hb-depleted CSF and Hb-restored CSF. (c) Dose-response curve for Hb with (data from Figure 4(a)) or without Hp. (d) The oxidative potential of patient CSF over time after aneurysmal subarachnoid hemorrhage (aSAH) with and without the addition of Hp or hemopexin (Hpx), as assessed by malondialdehyde (MDA) quantification with a thiobarbituric acid-reactive substances (TBARS) assay. The box in the boxplots bounds the interquartile range (IQR) divided by the median, while the whiskers extend to the highest and lowest value within the 1.5 × IQR, respectively.
Discussion
The main objective of this study was to investigate the clinical association of CSF-Hb with SAH-SBI and the diagnostic accuracy of daily CSF-Hb measurements for SAH-SBI. Based on a prospective cohort of 47 patients and 415 CSF samples, we provide very strong evidence for a positive association between CSF-Hb and the occurrence of SAH-SBI. In addition, CSF-Hb markedly exceeded the diagnostic accuracy of established prediction methods. Also, we aimed to rationalize the role of CSF-Hb as a targetable driver of SAH-SBI. We showed that, within a clinically relevant concentration range, CSF-Hb induced vasoconstriction and oxidized unsaturated lipids ex-vivo, suggesting that it might act as an upstream toxin. The Hb-scavenger haptoglobin and the heme-scavenger hemopexin effectively counteracted both toxicity mechanisms within the clinically relevant dose range. Collectively, the strong clinical association between high CSF-Hb levels and SAH-SBI, the underlying pathophysiological rationale and the favorable effects of haptoglobin and hemopexin position CSF-Hb as a highly attractive biomarker and potential drug target.
CSF-Hb as a biomarker for SAH-SBI
In this study, EVD sampling-based CSF-Hb showed a diagnostic accuracy for SAH-SBI that considerably exceeded that of existing methods. While often used in clinical practice, established clinical (WFNS, Hunt & Hess) and radiological (BNI, modified Fisher) scores show a limited predictive accuracy for SAH-SBI.25–28 The performance of combined scores or machine-learning-based models for predicting SAH-SBI is also only slightly better than that of the established clinical and radiological scores.29,30 The value of daily assessments of arterial flow velocity in the large cerebral arteries using TCD is controversial because of its limited diagnostic precision and high interrater variability31,32 and because it cannot be applied in one-fifth of patients due to anatomical reasons. 33 Due to the lack of superior alternatives, however, TCD is recommended by current guidelines and has been adopted by many clinical centers.11,34 In our cohort, the diagnostic accuracy of TCD for SAH-SBI was clearly below that of CSF-Hb. Moreover, literature-reported performance measures of proposed biomarkers for SAH-SBI are also substantially inferior to CSF-Hb.35,36 In addition to its high diagnostic accuracy for SAH-SBI, CSF-Hb is characterized by its simple and reliable analytics. Precise CSF-Hb values could be determined bedside with a simple two-step procedure consisting of a centrifugation step to remove cells and debris from the CSF and subsequent spectrophotometry with automated spectral deconvolution. Furthermore, spectrophotometry allows the absolute quantification of CSF-Hb without parallel measurements of calibration samples or to generate standard curves. Collectively, these advantages significantly reduce analytical costs and turnaround times and may enhance the widespread applicability and use of CSF-Hb quantification.
CSF-Hb as a pathophysiological driver of SAH-SBI
Despite the fact that Hb has long been considered a relevant pathophysiological factor in the development of SAH-SBI,7,13,14 only recently preclinical studies have experimentally determined that CSF-Hb induces pathophysiological processes reflecting specific features of SAH-SBI, such as vasospasm, lipid oxidation, and neuronal damage.15–17 However, a significant limitation of prior studies was that the explored Hb and heme concentrations were not based on a rational assessment of relevant CSF-Hb concentrations in patients. Indeed, our previous experiments in a sheep vasospasm model 15 defined effects of CSF-Hb at concentrations at least tenfold above the critical inflection point implied by our current patient data. Furthermore, our previous ex-vivo vasospasm model was incapable of defining physiologically relevant vasoconstrictive Hb concentrations because we used exogenous NO donors and, therefore, artificially set the concentration threshold at which Hb interrupted vasodilatory NO signaling. To address these issues, we redesigned our ex-vivo model to investigate how CSF-Hb interferes with an intrinsic vasodilator response. This was achieved by combining an algorithm-based prestretching protocol PGF2α as a pre-contracting agent, which induced endogenous, endothelial NO synthesis. With this new model, we confirmed that Hb is the major vasoconstrictor in patient CSF by comparing the vasoconstrictive effect of Hb-depleted and Hb-restored CSF. Additionally, we were able to determine that the active dynamic range of Hb concentrations in the ex-vivo vasospasm model overlaps with critical inflection points that determine TCD flow velocities in large cerebral arteries and the presence or absence of SAH-SBI in our patient cohort. Within the same clinically relevant concentration range, we found that CSF-Hb was highly active as a lipid oxidant, indicating oxidative Hb toxicity to contribute to nonischemic neuronal damage. We attributed the lipid oxidation activity in patient CSF to the downstream Hb metabolite heme. Its selective neutralization with hemopexin virtually blocked the formation of MDA, while haptoglobin partially inhibited MDA formation.
CSF proteome dynamics after aSAH
Although our study followed a strongly hypothesis-driven approach, we attempted to further assess the phenomenon of CSF-Hb accumulation and toxicity within the broader context of biological processes reflected by the temporal dynamics of the CSF proteome. Based on LC-MS/MS analysis of Hb-depleted CSF from the two weeks high-risk phase for SAH-SBI, we found the strongest signals for those proteins suggestive of RBC lysis and macrophage accumulation. RBC lysis was indicated by an increase in erythrocyte glycolytic and antioxidant enzymes (e.g., CA1, CA2, CAT, and ALDOA) over time, while lineage-specific soluble cell surface receptors (e.g., CD163, CD14, CSF1R and TIMP-1) indicated the rapid and sustained accumulation of macrophages. Prior studies have suggested that leptomeningeal macrophages with enhanced expression of the Hb scavenger receptor CD163 accumulate in the subarachnoid space after aSAH.37,38 In in-vitro studies and in hemolytic mice, macrophages exposed to damaged RBCs, Hb, or heme acquired an idiosyncratic phenotype supporting an adaptive response consisting of heme detoxification, iron sequestration, and inflammatory suppression, aiming at promoting hematoma resolution and wound healing.39–42 The time course of the macrophage protein signatures in our patients coincided well with the early peak in CSF bilirubin, which reflects metabolic heme degradation by heme oxygenase. 43 We found bilirubin to reach a plateau on days 3 to 4 after aSAH, followed by an increase in CSF biliverdin. This implies a saturation of the cumulative metabolic capacity of resident and recruited macrophages in the subarachnoid space at that time point (Figure 6). Therefore, the delayed increase in CSF-Hb concentrations to toxic levels appears to be the combined result of the accelerated lysis of RBCs and the saturated capacity of erythrophagocytic and Hb-clearing macrophages. The accumulation of RBC- and macrophage-proteins occurred even with the rapid depletion of plasma proteins, such as apolipoproteins, coagulation factors and haptoglobin, reflecting rapid protein clearance from the CSF during the initial days after aSAH.
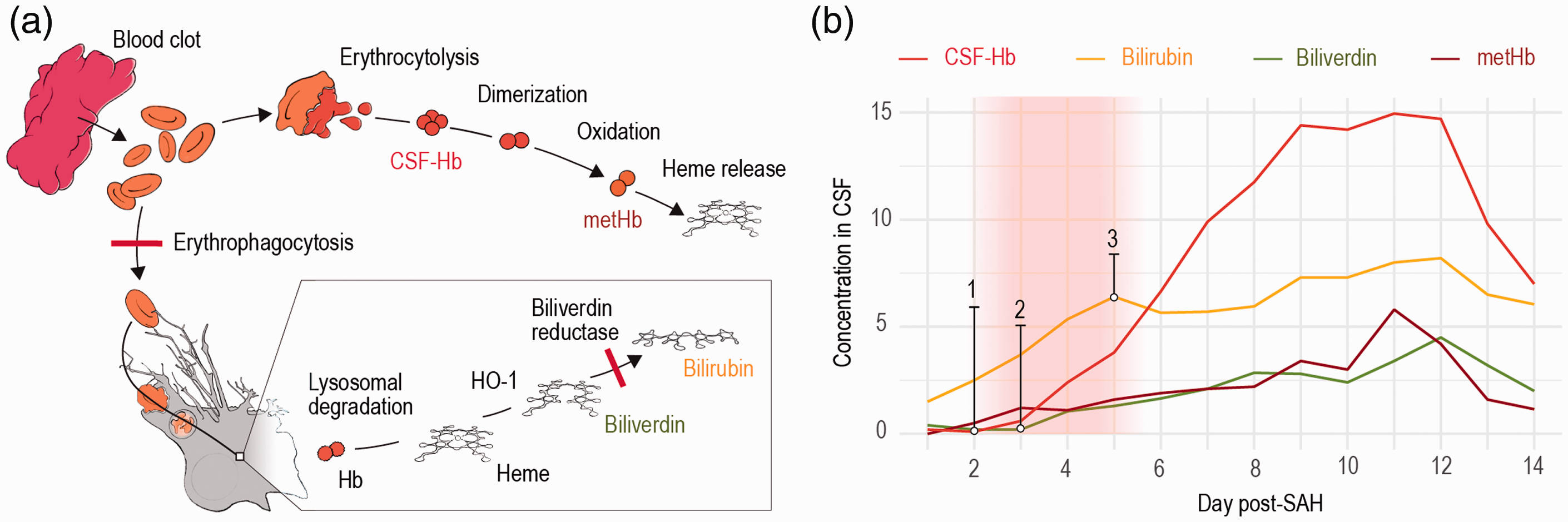
The pathophysiology of cerebrospinal fluid hemoglobin and heme metabolites after aneurysmal subarachnoid hemorrhage. (a) Schematic illustration of the pathophysiological processes in the cerebrospinal fluid (CSF) microenvironment after aneurysmal subarachnoid hemorrhage (aSAH). Erythrophagocytosis and the consecutive intracellular processes of hemoglobin (Hb) degradation and heme metabolization are shown at the bottom. The saturation of the biliverdin reductase and the erythrophagocytosis is indicated by red bars. Erythrocytolysis in the subarachnoid CSF space is shown at the top. Liberated Hb is oxidized to methemoglobin (metHb) before heme is released. (b) CSF concentrations of Hb (CSF-Hb), bilirubin, biliverdin and metHb over time with the assumed saturation period of biliverdin reductase and phagocytosis indicated by a red shaded area, resulting in an increase of CSF-Hb (1) and biliverdin (2) as well as the plateauing of bilirubin (3).
CSF-Hb as a therapeutic target for SAH-SBI
Our data reinforce the idea of CSF-Hb as an attractive drug target to prevent and treat SAH-SBI. This is supported by the strong clinical association between CSF-Hb and SAH-SBI, the underlying pathophysiological rationale behind this association, and the favorable functional effects of haptoglobin and hemopexin in neutralizing CSF-Hb in a clinically relevant concentration range for toxicity ex-vivo. Haptoglobin is part of the physiological scavenger system for extracellular Hb, providing the most upstream antagonization of Hb toxicity by preventing vascular and tissue translocation of the large Hb-haptoglobin complex, blocking heme release, stabilizing Hb radical reactions, and accelerating Hb clearance by the macrophage CD163 receptor pathway.20,44–46 In our CSF analysis, endogenous haptoglobin was shown to be cleared from the CSF within the first few days after aSAH before the subsequent release of relevant amounts of CSF-Hb. Thus, the protective function of endogenous haptoglobin seems negligible. In a previous animal study, intraventricularly administered haptoglobin inhibited Hb delocalization from the CSF into the brain parenchyma and smooth muscle cell layer of cerebral arteries, preventing Hb-induced aVSP. 15 Our findings support this protective function of haptoglobin by demonstrating its anti-vasoconstrictive and antioxidant effects at a clinically relevant Hb concentration range. The antioxidant function of the heme scavenger protein hemopexin even surpassed that of haptoglobin. This suggests that a fraction of heme had already been released from oxidized or degraded CSF-Hb, forming a pool of oxidative free heme that remained associated with low-affinity heme-binding proteins. Therefore, it appears reasonable that the therapeutic combination of intraventricularly administered haptoglobin and hemopexin may exert synergistic protection.
Limitations and pertinent research questions
While we believe that a clinical stratification of SAH-SBI into aVSP, DCI, and DIND is useful, it must be recognized that the underlying pathophysiology of secondary injury after aSAH is multifactorial and should not be arbitrarily simplified. While our ex-vivo assays cover specific aspects of dose-dependent CSF-Hb toxicity that correlate with aVSP, DCI, and DIND in patients, they cannot cover the entire spectrum of the complex pathophysiology of SAH-SBI. For instance, recent studies indicate that microthrombi formation after aSAH contributes to the development of DCI and DIND.47,48 CSF-Hb might be also involved in this, as endothelial NO scavenging by Hb has been shown to disinhibit platelet adhesion and aggregation.49–51 In addition, there may be an indirect link to Hb-induced microvasospasms,15,17,52–56 promoting the formation of microthrombi by resulting mechanical endothelial damage. Furthermore, there is recent preclinical evidence for acute and persistent loss of neurovascular coupling after aSAH, which may be pathophysiologically relevant both in the context of early brain injury as well as the development of SAH-SBI.9,57,58 Further studies are needed to determine the role of CSF-Hb in this context. Another relevant pathophysiological aspect, which is not covered by our ex-vivo assays, is the potential uptake of CSF-Hb by neurons via CD163 and associated neuronal damage.59–64 More investigations on the dose dependency of this effect and the controversial effect of haptoglobin are needed.65–67 Further, it must be emphasized that only indirect conclusions about erythrophagocytosis by macrophages based on the examination of the CSF proteome can be drawn in our study. No macrophages were isolated from the CSF. No conclusions can be drawn regarding the origin of the responsible macrophages, although a distinction between border-associated and peripheral macrophages in particular would be of pathophysiological and therapeutic interest. 68 In addition, it remains unclear how Hb is cleared from the CSF. Conceivable options are a clearing via meningeal lymphatic vessels or via the choroid plexus.69–72 After SAH, the absorptive function of these structures might be impaired. On the one hand, CSF-Hb may play an etiological role as a toxin in this functional impairment; on the other hand, its CSF concentration may be influenced by a reduced clearance via these structures. The fact that the CSF sampling in this study was conducted via an EVD located in the lateral ventricle may represent a certain limitation regarding some of the pathophysiological conclusions. The measured CSF-Hb concentrations represent local conditions in the lateral ventricle and do not necessarily correspond to the conditions in other CSF compartments (e.g., basal cisterns). Although, patients with IVH showed a pattern with an early peak and subsequent plateau, patients without IVH reached comparable CSF-Hb levels later on. This suggests the presence of a considerable redistribution of CSF-Hb across the whole CSF space, leading to a rather homogeneous distribution over time. A potential limitation of the clinical data results from the high number of missing observations in the daily assessment of DIND, DCI or aVSP. This is an intrinsic limitation in any study of aSAH patients, as the neurological assessment of patients with aSAH is frequently limited due to intubation and sedation, and cranial imaging is not conducted every day. The aim of using a strict definition of DIND, DCI or aVSP was to exclude uncertainties in the acquired data and to increase the reliability in the association between CSF-Hb and SAH-SBI. The homogeneous distribution of the measured CSF-Hb values within the NAs argues against a systematic error. In addition, our clinical study is limited by a selection of severe clinical cases due to the inherent need for an EVD. In our center, the indication for an EVD in a patient with aSAH is an impending acute hydrocephalus. Therefore, no conclusions can be made about patients without hydrocephalic congestion. Moreover, we cannot infer a potential relationship between CSF-Hb and acute hydrocephalus. In order to answer this question, a comparable study would have to be performed in centers where also patients without acute hydrocephalus receive EVD, e.g. to monitor intracranial pressure. Furthermore, this study is limited by the number of patients. While there is an adequate statistical power for conclusions regarding the association of daily CSF-Hb with both baseline and outcome parameters (per day and per patient), this does not apply to conclusions regarding the association of patient-specific baseline and outcome characteristics, e.g. the association between gender and the occurrence of SAH-SBI.73,74 Finally, this is an exploratory single center study intended to provide a solid basis for a preregistered multicenter validation study.
Conclusions
In this study, we provide very strong evidence for a positive association between CSF-Hb and the occurrence of SAH-SBI. Furthermore, CSF-Hb was shown to exhibit a high diagnostic accuracy for SAH-SBI. Within a clinically relevant concentration range, CSF-Hb induced vasoconstriction and oxidized unsaturated lipids ex-vivo, suggesting that it acts as an upstream toxin released from the subarachnoid hematoma. Based on these clinical and ex-vivo data, we provide a plausible pathophysiological link between the initial blood volume, the generation of toxic CSF-Hb and the development of SAH-SBI. The Hb-scavenger haptoglobin and the heme-scavenger hemopexin effectively counteracted both toxicity mechanisms within the clinically relevant dose range ex-vivo. Collectively, these findings position CSF-Hb as a highly attractive biomarker and potential drug target.
Supplemental Material
sj-pdf-1-jcb-10.1177_0271678X211020629 - Supplemental material for Cerebrospinal fluid hemoglobin drives subarachnoid hemorrhage-related secondary brain injury
Supplemental material, sj-pdf-1-jcb-10.1177_0271678X211020629 for Cerebrospinal fluid hemoglobin drives subarachnoid hemorrhage-related secondary brain injury by Kevin Akeret, Raphael M Buzzi, Christian A Schaer, Bart R Thomson, Florence Vallelian, Sophie Wang, Jan Willms, Martina Sebök, Ulrike Held, Jeremy W Deuel, Rok Humar, Luca Regli, Emanuela Keller, Michael Hugelshofer and Dominik J Schaer in Journal of Cerebral Blood Flow & Metabolism
Footnotes
Funding
Acknowledgments
Declaration of conflicting interests
Authors’ contributions
Disclosure
ORCID iDs
Supplemental material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.