Ochnaceae DC. includes more than 600 species that exhibit potential values for environmental ecology, ornamental, pharmaceutical, and timber industries. Although studies on phylogeny and phytochemicals have been intensively conducted, chloroplast genome data of Ochnaceae species have not been fully explored. In this study, the next-generation sequencing method was used to sequence the chloroplast genomes of Ochna integerrima and Ochna serrulata which were 157 329 and 157 835 bp in length, respectively. These chloroplast genomes had a quadripartite structure and contained 78 protein-coding genes, 30 tRNAs, and 4 rRNAs. Comparative analysis revealed 8 hypervariable regions, including trnK_UUU-trnQ_UUG, rpoB-psbM, trnS_GGA-rps4, accD-psaI, rpl33-rps18, rpl14-rpl16, ndhF-trnL_UAG, and rps15-ycf1 among 6 Ochnaceae taxa. Additionally, there were shared and unique repeats among 6 examined chloroplast genomes. The notable changes were the loss of rpl32 in Ochna species and the deletion of rps16 exon 2 in O. integerrima compared to other taxa. This study is the first comprehensive comparative genomic analysis of complete chloroplast genomes of Ochna species and related taxa in Ochnaceae. Consequently, the current study provides initial results for further research on genomic evolution, population genetics, and developing molecular markers in Ochnaceae and related taxa.
Ochnaceae DC. contains 36 genera of 638 species distributed in the tropical region.1Ochna, the second-largest genus of Ochnaceae, contains 79 species which are distributed in tropical regions of Africa and Asia.1 New species of Ochnaceae have been also reported recently.2-4 Previously, morphologies were the main topics of study in Ochnaceae that assisted in classification.5-7 Moreover, biogeography of Quiinoideae, an Ochnaceae subfamily, and divergence time for Ochnaceae were conducted.8,9 For phylogenetic analysis, different data were employed, such as nuclear and chloroplast genomes.10-12 Recently, phylogenetic relationships and new infrageneric classification of Ochnaceae have been proposed based on nuclear genes and exon data.13,14 These studies provide the most comprehensive classification of Ochnaceae up-to-date.
In addition to phylogenetic studies, the medicinal component of Ochnaceae species has also been explored. Different chemical compositions have been screened in Lophira and Ochna species.15-18 These phytochemical analyses revealed antioxidant, anti-inflammatory, antibacterial, antiviral, and antiproliferative features. In Ochna taxa, oils and ochnaflavone were detected in Ochna afzelii R.Br. ex Oliv., Ochna serrulata (Hochst.) Walp., and Ochna pretoriensis E.Phillips. These results revealed the potential of Ochnaceae members, especially Lophira and Ochna, as a new source for medicinal production.
Chloroplast is vital to most land plants due to its function for performing photosynthesis.19 Chloroplast contains genetic materials called chloroplast genomes (cpDNA), which have genes related to photosynthesis. cpDNA is a circular quadripartite molecule composed of a large single copy (LSC), a small single copy (SSC), and 2 inverted repeat regions (IRs).20 Together with the nuclear and the mitochondrial genomes, the cpDNAs store the traces of the evolutionary history of land plants through various genomic events (ie, gene deletion, duplication, and pseudogenization). For example, deletion of one IR region in cpDNA was found in gymnosperms and legumes.21,22 Gene deletion or pseudogenization was more common in heterotrophic species such as parasite and mycoheterotrophic plants.23,24 Additionally, sequence data of cpDNA were applied to reconstruct the phylogeny of land plants.25 These studies revealed the value of cpDNA in elucidating the evolution of plants. In Ochnaceae, chloroplast genome sequences of Testulea gabonensis Pellegr. (GenBank accession MZ274137), Lophira lanceolata Tiegh. ex Keay (MZ274136), Lophira alata Banks ex C.F.Gaertn. (MZ274135), and Sauvagesia rhodoleuca (Diels) M.C.E.Amaral (MW772237) were reported.26,27 Although studies on phylogeny and medicinal features among Ochnaceae were intensively conducted, only 4 out of 638 species had records for complete cpDNA, which is insufficient to elucidate the evolutionary history of Ochnaceae and Malpighiales in general. Notably, there is no record of complete chloroplast genome in 2 largest genera including Ouratea and Ochna of Ochnaceae that might contain different genomic events (ie, gene deletion, pseudogenization, and duplication) during the evolutionary history.
Therefore, in this study, the Illumina sequencing was used to obtain the entire chloroplast genomes of Ochna integerrima (Lour.) Merr. and Ochna serrulata. The newly sequenced chloroplast genomes together with the 4 previously published ones of Ochnaceae were used to (1) investigate genomic evolution regarding gene content and order, (2) locate highly variable regions among cpDNA of Ochnaceae, and (3) characterize repeats in 6 available cpDNA of Ochnaceae. This study provides the first comprehensive comparative genomics analysis of the complete chloroplast genomes which are essential for further studies on evolutionary history, molecular marker development, and population genetics in Ochnaceae.
Materials and Methods
Plant materials and DNA extraction
Fresh leaves of Ochna integerrima and Ochna serrulata were collected at Can Tho City, Vietnam (N 9°58′19.423′E105°44′ 24.011′; Figure 1). There is no specific permission required for collecting Ochna species in Vietnam. The leaves were dried in silica gel until the total genomic DNA extraction step which was done using DNeasy Plant Mini Kit (Qiagen, Germany). The quality of extracted DNA was checked using gel electrophoresis and the Nanodrop One spectrophotometer (ThermoFisher, USA). The samples which exhibited clear bands on agarose gel and showed a concentration of ~100 ng/μL (with a 260/280 ratio of ~1.8 and 260/230 ratio of 2.0-2.2) were further processed to the next step.
Flower of Ochna species: (A) flower of Ochna integerrima and (B) flower of Ochna serrulata.
Genome sequencing and assembly
The high-quality DNA samples of O. integerrima and O. serrulata were used to prepare the sequencing library using the Ultra II DNA Library Prep Kit for Illumina (NEB, UK). The library preparation was processed in accordance with the protocol of the manufacturer and sequencing was conducted using the Illumina NextSeq 1000 system with a 150 bp paired-end reading. Raw data of 3.361.080 reads and 3.926.308 reads were generated for O. integerrima and O. serrulata, respectively. The obtained raw reads were quantified by FastQC28 and the low-quality reads (containing N bases and Qscore ⩽20) were filtered using the Trimmomatic tool.29 The filtered reads were then assembled to obtain chloroplast genome by NOVOPlasty software (version 4.3.1) with the chloroplast genome of Lophira alata as the reference.30 The newly completed cpDNA sequences were annotated using GeSeq.31 The gene contents were manually checked by Geneious Prime v2022.1 to confirm the start and stop codons of each gene (https://www.geneious.com). Additionally, tRNAScan-SE was used to validate the tRNA sequences.32,33 The complete cpDNA sequences of O. integerrima and O. serrulata were deposited to GenBank with accession numbers OQ130028 and OQ130029, respectively. The maps of cpDNA were illustrated using OGDRAW program.34
Comparative genomic analysis
The complete cpDNAs of 4 species in Ochnaceae were downloaded from NCBI, including Sauvagesia rhodoleuca (MW772237), Testulea gabonensis (MZ274137), Lophira lanceolata (MZ274136), and Lophira alata (MZ274135). The total of 6 cpDNA sequences of Ochnaceae (the above mentioned species and 2 from this study) were imported to Geneious Prime to extract the information on length, GC content (%), junctions among LSC, SSC, and IR regions, and the number of genes. MAUVE embedded in Geneious Prime was used to align 6 cpDNAs.35
The aligned sequences were used to calculate nucleotide diversity (Pi value) with a window length of 2000 sites and step size of 200 sites using DnaSP 6.36 Phobos program in Geneious Prime was used to identify simple sequence repeats (SSRs) with categories of mononucleotide (at least 10 bp in length), dinucleotide (at least 12 bp in length), trinucleotide (at least 15 bp in length), tetranucleotide (at least 16 bp in length), pentanucleotide (at least 20 bp in length), and hexanucleotide (at least 24 bp in length) repeats.37 REPuter program was used to locate long repeats (at least 20 bp in length) which include forward, reverse, complement, and palindromic types.38 All repeats were checked for length and location in Geneious Prime.
Phylogenetic analysis
The complete chloroplast genomes of Ochnaceae species and related taxa were downloaded from NCBI (Supplemental Table S1). The cpDNA genome of Galphinia angustifolia (Malpighiaceae) was used as an outgroup. A total of 73 protein-coding regions (except ycf1, ycf2, rpl23, rpl32, rps16, and infA because of gene loss and pseudogenization among surveyed chloroplast genomes) were aligned to make a data matrix using MUSCLE program.39 The jModelTest 2.1.10 program was used to identify the best model for the data matrix.40 Consequently, GTR+I+G model with Akaike information criterion was selected for the data matrix to reconstruct phylogenetic relationships between Ochnaceae and related taxa using Maximum Likelihood (ML) and Bayesian Inference (BI) methods. For the ML method, the IQ-TREE 2 server was employed with 1000 bootstrap replications.41 Meanwhile, MrBayes v3.2.7 program was used for the Bayesian Inference method with 1,000,000 generations that resulted in a split frequency of less than 0.01.42 Then, a total of 25% of the samples were discarded for BI method. The phylogenetic trees were illustrated and modified using Figtree v1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/).
Results
Feature of chloroplast genomes in Ochnaceae
In Ochnaceae, the length of available cpDNA ranged from 157 300 bp to 159 925 bp, in which L. alata had the largest size of 159 925 bp. In our study, the chloroplast genomes of O. integerrima and O. serrulata were 157 329 bp and 157 835 bp in length, respectively (Figure 2, Table 1). The overall GC contents of Ochna species (36.7%) were slightly higher than other species including S. rhodoleuca (36.4%), T. gabonensis (36.5%), and L. alata and L. lanceolata (36.4%). The lengths of LSC, IR, and SSC regions varied among examined species (Table 1). There were 79 protein-coding genes, 30 tRNAs, and 4 rRNAs in cpDNA of S. rhodoleuca, T. gabonensis, L. alata, and L. lanceolata (Tables 1 and 2). Our chloroplast genome annotation showed that the same numbers of tRNAs and rRNAs were found in the 2 Ochna cpDNAs; however, they only contain 78 protein-coding genes (Figure 2). The absence of functional gene resulted from the deletion of rpl32 gene which located between ndhF and trnL-UAG in SSC region of Ochna cpDNAs. Another remarkable feature was a deletion of the second exon of rps16 in O. integerrima, causing the pseudogenization of the rps16.
Map of the chloroplast genome of Ochna integerrima and O. serrulata. Genes positioning outside the circle are transcribed clockwise, whereas genes found inside the circle are transcribed counterclockwise. Genes with different functional groups are colored. LSC, large single copy; SSC, small single copy; IRA-IRB, inverted repeat regions.
Features of chloroplast genomes among surveyed species of Ochnaceae.
Species
Sauvagesia rhodoleuca
Testulea gabonensis
Lophira lanceolata
Lophira alata
Ochna serulata
Ochna integerrima
Accession number
MW772237
MZ274137
MZ274136
MZ274135
OQ130029
OQ130028
Size (bp)
157 300
157 502
159 869
159 925
157 835
157 329
Overall %GC
36.4
36.5
36.4
36.4
36.7
36.7
LSC length (bp)
86 021
85 248
88 064
88 096
88 123
87 639
% GC in LSC
34
34.4
34.2
34.2
34.4
34.4
IR length (bp)
26 571
26 567
26 446
26 456
26 074
26 074
% GC in LSC
42.5
42.4
42.5
42.5
42.6
42.7
SSC length (bp)
18 137
19 120
18 913
18 917
17 564
17 542
% GC in LSC
30.2
29.9
30.1
30.1
30.5
30.6
Protein-coding genes
79
79
79
79
78
78
rRNA
30
30
30
30
30
30
tRNA
4
4
4
4
4
4
Gene content of chloroplast genomes among Ochnaceae species.
, genes with introns; Ψ, pseudogene only in O. integerrima; §, duplication in S. rhodoleuca.
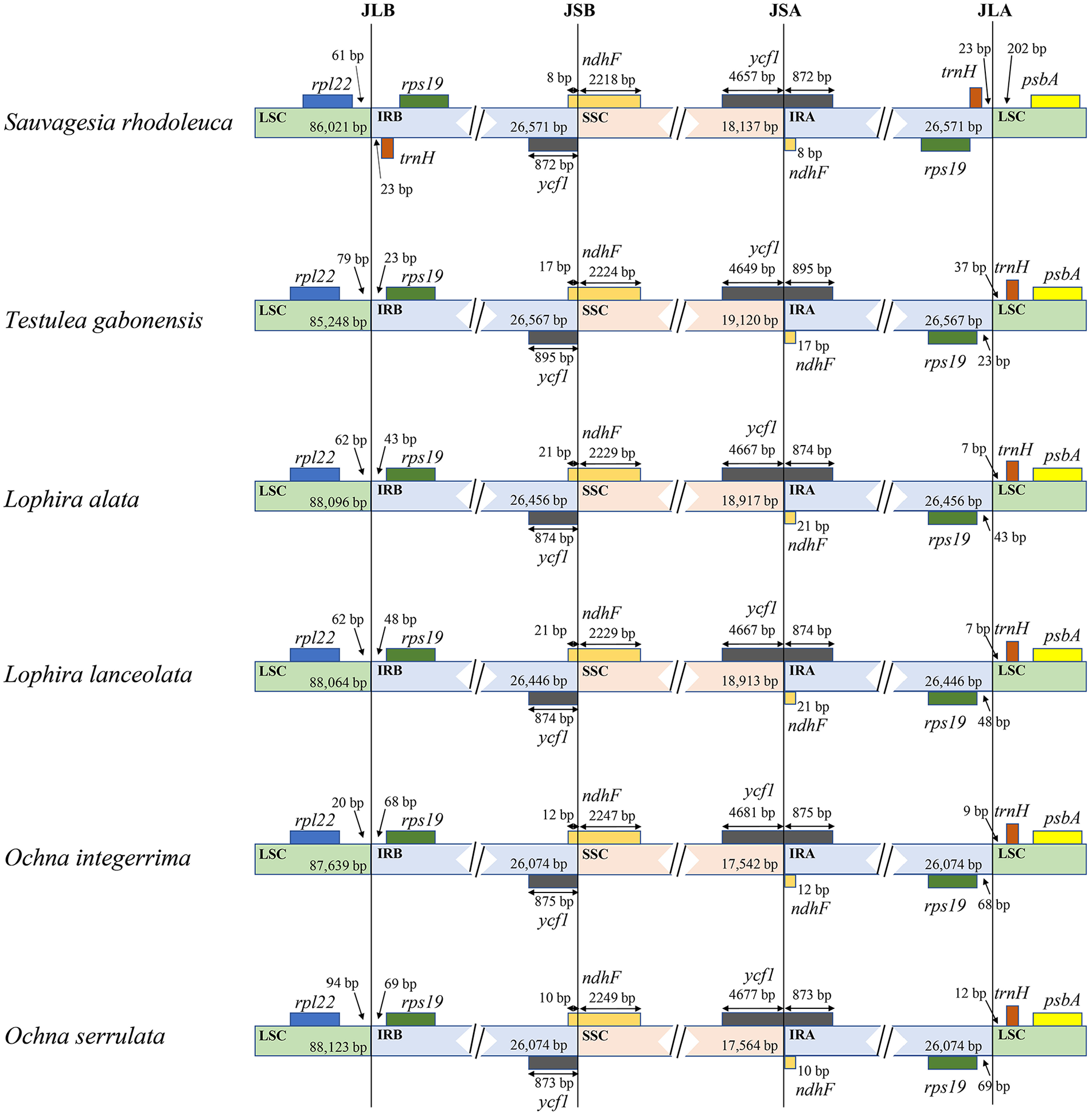
Albeit the lengths of cpDNA varied in Ochnaceae, the contents of junctions among LSC, SSC, and IR regions were relatively constant (Figure 3). Specifically, the junction between SSC and IR regions located in the coding region of ndhF and ycf1 in all examined taxa. A part of ycf1 (ranging from 872 to 875 bp) and ndhF (ranging from 8 to 21 bp) were found in IR regions. Notable, the IR regions in S. rhodoleuca extended to include trnH_GUG, resulting in duplication of trnH_GUG.
Junctions comparison among large-single, small-single, and inverted repeat regions in 6 Ochnaceae species. JSA: junction between LSC and IRA. JSA: junction between SSC and IRA. JSB: junction between LSC and IRB. JSB: junction between SSC and IRB. The numbers outside the boxes indicate the base pair distance between annotated regions. The distances are not drawn in proportion.
We calculated the value of nucleotide variability (Pi) and revealed that more highly variable regions were found in LSC and SSC than those in IR (Figure 4). There were 8 variable regions including trnK_UUU-trnQ_UUG, rpoB-psbM, trnS_GGA-rps4, accD-psaI, rpl33-rps18, rpl14-rpl16, ndhF-trnL_UAG and rps15-ycf1, which had high Pi value (Pi > .04). Two regions of trnK_UUU-trnQ_UUG and rpl14-rpl16 exhibited the tops of variability (Pi > .06) in 6 Ochnaceae species.
Nucleotide diversity of 6 chloroplast genomes in Ochnaceae.
Composition of repeats in six chloroplast genomes
The simple sequence repeat analysis revealed fewer repeats present in the cpDNA of O. integerrima (63 repeats) and O. serrulata (62 repeats) than in other taxa which contained 73 to 98 repeats in their cpDNAs (Figure 5A, Supplemental Table S2). Among 6 categories of SSRs, there were no records for penta- and hexanucleotide repeats in cpDNA of all surveyed species. Two tetranucleotide repeats were present only in S. rhodoleuca, which were 16 bp in length. Trinucleotide repeats were found in 4 out of 6 examined taxa, of which the longest length (24 bp) was detected in L. alata. The mono- and dinucleotide repeats were found in cpDNAs of all 6 taxa, of which the longest lengths were 25 and 42 bp, respectively. The largest mononucleotide repeat was found in psbE-petL region of T. gabonensis cpDNA, which also contained the largest trinucleotide repeat in ndhF-rpl32 intergenic space (Supplemental Table S2). Most of SSRs were composed of A and T nucleotides and located in the non-coding regions.
Features of repeats in 6 Ochnaceae taxa: (A) information of single sequence repeats (SSRs) and (B) comparison of long repeats.
Similar to SSRs, long repeats in cpDNAs (ranging from 20 to 42 bp) were located mainly in non-coding regions (Figure 5B, Supplemental Table S3). Among 4 examined repeat types including forward, reverse, complement and palindromic types, only forward and palindromic repeats were found in 6 Ochnaceae taxa. Additionally, forward repeats were more abundant than the palindromic counterparts. In 6 surveyed species, T. gabonensis had the highest number of repeats (30 records) whereas the lowest number of repeats was found in O. integerrima (13 repeats). Among repeats, there were 22 unique ones in cpDNA of T. gabonensis, followed by S. rhodoleuca which had 19 unique repeats (Supplemental Table S3). Only 3 unique repeats were found in L. lanceolata and O. integerrima. In contrast, there were 5 shared repeats among 6 surveyed taxa, located in trnS_GCU, trnS_UGA,trnS_GGA, psaA, psaB, psbT-psbN intergenic space, ndhA intron, and rps12-trnV_GAC intergenic space. In Lophira species, there were 5 shared repeats whereas only 3 shared repeats were found in Ochna taxa (Supplemental Table S3).
Phylogenetic relationships between Ochna members and related taxa
The Maximum Likelihood (ML) and Bayesian Inference (BI) analyses resulted in the same topology of phylogenetic trees (Figure 6). The monophyly of Ochnaceae was highly supported (BS = 100 and PP = 1). Within the Ochnaceae family, Ochna species was close to those of Lophira. Meanwhile, Testulea gabonensis was sister to all other surveyed Ochnaceae taxa. Furthermore, highly supported monophyly of Clusiaceae, Calophyllaceae, Hypericaceae, Podostemaceae, and Rhizophoraceae families were found (Figure 6).
The phylogenetic tree of Ochnaceae and related species inferred from 73 protein-coding sequences in the chloroplast genomes using Maximum likelihood and Bayesian inference methods. The bold names indicate newly generated chloroplast genome in this study. The numbers indicate posterior probability (PP) and bootstrap (BS) values. Only values of PP < 1 and BS < 100 were shown on the phylogenetic tree. The names on the right side represent family names of the samples.
Discussion
The newly sequenced cpDNAs of Ochna species have a similar structure to the published relatives in Ochnaceae (Figures 2 and 3, Table 1). However, 8 hypervariable regions were found in 6 examined species (Figure 4). Previously, hypermutable areas in cpDNA were identified among land plants that had specific variable regions.43,44 A notable change in cpDNAs of Ochnaceae was the expansion of IR region that included trnH_GUG in S. rhodoleuca (Figure 3). Contraction and expansion of IR regions resulted in different sizes and gene contents of cpDNA.45,46 The junctions among LSC, SSC, and IR regions were previously classified into 5 types based on the gene content in Liliales.47 However, the genomic data of heterotrophic species revealed complex characteristics of LSC/SSC/IR junctions, and no quadripartite structure was also recorded.48-50 Therefore, it is difficult to identify a common trend of LSC/SSC/IR boundaries among angiosperms. In this study, although LSC/SSC/IR junctions were relatively stable in the intergenic space between rpl22-rps19, the shift of trnH_GUG to IR regions of S. rhodoleuca revealed a possibility of structural changes in cpDNA of Ochnaceae members. Therefore, more complete cpDNAs of Ochnaceae should be conducted to understand an overview of the evolution of LSC/SSC/IR boundaries.
In cpDNA, SSRs and long repeats were valuable tools for studies on genetic diversity, evolutionary history, hybridization, and molecular markers among land plants.51,52 In 6 surveyed Ochnaceae species, SSRs and long repeats were located across cpDNAs non-coding and coding regions (Supplemental Tables S2 and S3). Besides shared repeats among Ochnaceae, there were unique repeats in each of the surveyed taxa. These unique repeats are helpful information for developing molecular markers and studying the evolutionary history of Ochnaceae species.
Gene loss in cpDNA was common in heterotrophic plants because genes related to photosynthesis are not necessary.53,54 However, in autotrophic plants, loss of genes seldom occurs. Previously, deletions of some genes such as rps16, infA, rpl32, and ycf4 were identified in various species.54-57 The loss of genes in cpDNA was replaced by homologs that were transferred to the nuclear genome.55,58-60 The reason for gene loss could be the presence of hypervariable regions. For example, in the cpDNA of Lathyrus odoratus L., ycf4 was lost, and there was a region in which the mutation rate was 20 times higher than related taxa.61 This higher rate could be the possible explanation for the loss of ycf4 in L. odoratus. In the current study, results of nucleotide diversity revealed higher Pi values in the regions containing rps16 and rpl32 compared to other areas in cpDNA of Ochnaceae (Figure 4). Additionally, trnK_UUU – trnQ_UUG intergenic space had SSRs in examined species (Supplemental Tables S2 and S3). These features in cpDNA suggested a high possibility of genomic events regarding gene content, complete particularly deletion of rpl32 and partial loss of rps16 in Ochnaceae. Specifically, rpl32 was lost in O. integerrima and O. serrulata whereas exon 2 of rps16 was not found in O. integerrima (Figure 2, Tables 1 and 2). The loss of rps16 and rpl32 were previously reported in Populus and Euphorbia species.55,56 Additionally, continuous deletion of rps16 regions was found in cpDNA of Melanthiaceae members.62 In 2 Ochna species, loss of rps16 exon 2 was found in O. integerrima, whereas full rps16 was intact in O. serrulata, suggesting different stages of rps16 loss in Ochna as well as related taxa in Ochnaceae. Previously, a comparative analysis of 18 complete cpDNAs of Passiflora (Passifloraceae, Malpighiales) revealed the loss and pseudogenization of various genes (ie, accD, rpl20, rpl22, rps7, rps16, rpoA, ycf1, and ycf2) as well as deletion of one IR region.54
In previous phylogenetic studies, partial chloroplast genomes and 353 nuclear genes resulted in a highly supported backbone phylogenetic tree of Ochnaceae including Medusagynoideae, Quiinoideae, and Ochnoideae subfamilies.8,10,11,13 Although the monophyly of most genera of Ochnaceae has been revealed, polyphyletic status of Sauvagesia and Campylospermum need further investigation.10 Additionally, the phylogenetic relationships at the infrageneric level witnessed low support, suggesting that more genomic data should be employed for phylogenetic studies of Ochnaceae.11 In the current study, although the data of 73 protein-coding regions revealed high support within Ochnaceae, the low number of samples did not reflect the overall relationships (Figure 6). In Ochnaceae, there were only 6 complete cpDNA sequences among 638 species in this current study. Therefore, more genomic data should be obtained to explore the overview of evolutionary history, especially gene loss in cpDNA and infrageneric relationships in the Ochnaceae family.
Conclusions
This study provided the first comparative genomic result of complete chloroplast genomes in Ochnaceae that includes potential taxa with ornamental values, the pharmaceutical and timber industry, and environmental ecology. The results revealed a stable trend in the structure of chloroplast genomes of surveyed Ochnaceae taxa. However, 8 hypervariable regions were identified among 6 surveyed Ochnaceae species, which can be applied for reconstructing infrageneric relationships in Ochnaceae. Additionally, the partial deletion of rps16 and complete loss of rpl32 in Ochna species suggested various potential genomic events in Ochnaceae that might correlate with speciation in Ochnaceae. Therefore, complete chloroplast genomes will provide essential data for further research on genomics, evolutionary history, and genetic populations in Ochnaceae and related taxa.
Supplemental Material
sj-docx-1-evb-10.1177_11769343231210756 – Supplemental material for New Insights Into The Evolution of Chloroplast Genomes in Ochna Species (Ochnaceae, Malpighiales)
Supplemental material, sj-docx-1-evb-10.1177_11769343231210756 for New Insights Into The Evolution of Chloroplast Genomes in Ochna Species (Ochnaceae, Malpighiales) by Nguyen Nhat Nam, Nguyen Hoang Danh, Vu Minh Thiet and Hoang Dang Khoa Do in Evolutionary Bioinformatics
Footnotes
Author Contributions
Conceptualization,N.N.N.,V.M.T and H.D.K.D.;collected data,N.N.N and N.H.D.;formal analysis,N.N.N. and H.D.K.D.;writing—original draft preparation,N.N.N and N.H.D.;writing—review and editing,V.M.T and H.D.K.D.;All authors have read and agreed to the published version of the manuscript.
Funding:
The author(s) disclosed receipt of the following financial support for the research,authorship,and/or publication of this article: This research was fully supported by Tra Vinh University under grant contract number 220/2022/HĐ.HĐKH&ĐT-ĐHTV. The author extends their appreciation to Nguyen Tat Thanh University for providing facilities to conduct this research.
Declaration of conflicting interests:
The author declared no potential conflicts of interest with respect to the research,authorship,and/or publication of this article.
ORCID iD
Hoang Dang Khoa Do
Data Availability
The data that support the findings of this study are available in the NCBI GenBank at ,reference numbers ( Ochna integerrima OQ130028;Ochna serrulata OQ130029).
Supplemental Material
Supplemental material for this article is available online.
CardosoDBOSHarleyRM.Sauvagesia paganuccii (Ochnaceae), a new species endemic to campo rupestre vegetation of Bahia, Brazil. Syst Bot. 2015;40:776-781.
3.
ShahTBurrowsJDarbyshireI.A new species of Ochna (Ochnaceae) from the Barberton Mountains of Mpumalanga, South Africa. Phytotaxa. 2018;374:241.
4.
BalkwillK.Ochna maguirei (Ochnaceae), a new species with corky bark from northern South Africa. S Afr J Bot. 2020;133:298-306.
5.
FurnessCA.Evolution of pollen and tapetal characters in Ochnaceae (Malpighiales). Int J Plant Sci. 2013;174:1134-1152.
6.
ReinalesSParra-OC.Phylogenetic position and evolution of glandular structures of the unusual and narrowly distributed genus Rhytidanthera (Ochnaceae). Bot J Linn Soc. 2020;193:84-99.
7.
DickoANattaAKBiaouHSSAkossouA.Assessing morphological traits variation and fruit production of Lophira lanceolata (Ochnaceae) in Benin. Am J Plant Sci. 2019;10:1048-1060.
8.
SchneiderJVZizkaG. Phylogeny, taxonomy and biogeography of Neotropical Quiinoideae (Ochnaceae s.l). Taxon. 2017;66:855-867.
9.
SchneiderJVJungcurtTCardosoD, et al. Predominantly eastward long-distance dispersal in pantropical Ochnaceae inferred from ancestral range estimation and phylogenomics. Front Ecol Evol. 2022;10:813336. doi:10.3389/fevo.2022.813336
10.
ShahTSchneiderJVZizkaG, et al. Joining forces in Ochnaceae phylogenomics: a tale of two targeted sequencing probe kits. Am J Bot. 2021;108:1201-1216.
11.
SchneiderJVPauleJJungcurtT, et al. Resolving recalcitrant clades in the Pantropical Ochnaceae: Insights from comparative phylogenomics of plastome and nuclear genomic data derived from targeted sequencing. Front Plant Sci. 2021;12:638650. doi:10.3389/fpls.2021.638650
12.
SchneiderJVSwensonUSamuelRStuessyTZizkaG.Phylogenetics of Quiinaceae (Malpighiales): evidence from trnL-trnF sequence data and morphology. Plant Syst Evol. 2006;257:189-203.
13.
ShahTMashimbaFHSuleimanHO, et al. Phylogenetics of Ochna (Ochnaceae) and a new infrageneric classification. Bot J Linn Soc. 2022;198:361-381.
14.
SchneiderJVJungcurtTCardosoD, et al. Phylogenomics of the tropical plant family Ochnaceae using targeted enrichment of nuclear genes and 250+ taxa. Taxon. 2021;70:48-71.
15.
Ngoua-Meye-MissoR-LSima-ObiangCNdongJDLC, et al. Phytochemical screening, antioxidant, anti-inflammatory and antiangiogenic activities of Lophira procera A. Chev. (Ochnaceae) medicinal plant from Gabon. Egypt J Basic Appl Sci. 2018;5:80-86.
16.
KalmobéJNdjonkaDBoursouDVildinaJDLiebauE.Phytochemical analysis and in vitro anthelmintic activity of Lophira lanceolata (Ochnaceae) on the bovine parasite Onchocerca ochengi and on drug resistant strains of the free-living nematode Caenorhabditis elegans. BMC Complement Altern Med. 2017;17:404.
17.
MöllerMSuschkeUNolkemperS, et al. Antibacterial, antiviral, antiproliferative and apoptosis-inducing properties of Brackenridgea zanguebarica (Ochnaceae). J Pharm Pharmacol . 2010;58:1131-1138.
18.
MakhafolaTJMcGawLJEloffJN.In vitro cytotoxicity and genotoxicity of five Ochna species (Ochnaceae) with excellent antibacterial activity. S Afr J Bot. 2014;91:9-13.
19.
HoweCJBarbrookACKoumandouVL, et al. Evolution of the chloroplast genome. Philos Trans R Soc Lond Ser B Biol Sci. 2003;358:99-107.
20.
DaniellHLinC-SYuMChangWJ.Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol. 2016;17:134.
21.
ChenCXiaXPengJWangD.Comparative analyses of six complete chloroplast genomes from the genus Cupressus and Juniperus (Cupressaceae). Gene. 2022;837:146696.
22.
ChoiI-SJansenRRuhlmanT.Lost and found: Return of the inverted repeat in the legume clade defined by its absence. Genome Biol Evol. 2019;11:1321-1333.
23.
FraileyDCChaluvadiSRVaughnJNCoatneyCGBennetzenJL.Gene loss and genome rearrangement in the plastids of five hemiparasites in the family Orobanchaceae. BMC Plant Biol. 2018;18:30.
24.
KimY-KJoSCheonS-H, et al. Extensive losses of photosynthesis genes in the plastome of a mycoheterotrophic Orchid, Cyrtosia septentrionalis (Vanilloideae: Orchidaceae). Genome Biol Evol. 2019;11:565-571.
25.
GitzendannerMASoltisPSWongGKRuhfelBRSoltisDE.Plastid phylogenomic analysis of green plants: a billion years of evolutionary history. Am J Bot. 2018;105:291-301.
26.
XieXHuangJZhangYZhuS.The complete chloroplast genome of Sauvagesia rhodoleuca, an endangered species endemic to China. Mitochondrial DNA B. 2021;6:2398-2399.
27.
MascarelloMAmalfiMAsselmanP, et al. Genome skimming reveals novel plastid markers for the molecular identification of illegally logged African timber species. PLoS One. 2021;16(6):e0251655.
BolgerAMLohseMUsadelB.Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114-2120.
30.
DierckxsensNMardulynPSmitsG.NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017;45(4): e18.
31.
TillichMLehwarkPPellizzerT, et al. Geseq - versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017;45(W11):W6-W11.
32.
LoweTMChanPP.tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016;44:W54-W57.
33.
ChanPLoweT.tRNAscan-SE: searching for tRNA genes in genomic sequences. Methods Mol Biol. 2019;1962:1-14.
34.
GreinerSLehwarkPBockR.OrganellarGenomeDRAW (OGDRAW) version 1.3.1: expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019;47:W59-W64.
35.
DarlingACMauBBlattnerFRPernaNT.Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004;14:1394-1403.
36.
RozasJFerrer-MataASánchez-DelBarrioJC, et al. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol. 2017;34:3299-3302.
37.
KraemerLBeszteriBGäbler-SchwarzS, et al. STAMP: extensions to the STADEN sequence analysis package for high throughput interactive microsatellite marker design. BMC Bioinformatics. 2009;10:41.
38.
KurtzSChoudhuriJVOhlebuschE, et al. REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001;29:4633-4642.
39.
EdgarRC.MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113.
40.
DarribaDTaboadaGLDoalloRPosadaD.jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 2012;9:772-772.
41.
MinhBQSchmidtHAChernomorO, et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the Genomic Era. Mol Biol Evol. 2020;37:1530-1534.
42.
RonquistFTeslenkoMvan der MarkP, et al. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61:539-542.
43.
WenFWuXLiT, et al. The complete chloroplast genome of Stauntonia chinensis and compared analysis revealed adaptive evolution of subfamily Lardizabaloideae species in China. BMC Genomics. 2021;22:161.
44.
YuJFuJFangYXiangJDongH.Complete chloroplast genomes of Rubus species (Rosaceae) and comparative analysis within the genus. BMC Genomics. 2022;23:32.
45.
PlunkettGMDownieSR.Expansion and contraction of the chloroplast inverted repeat in Apiaceae subfamily Apioideae. Syst Bot. 2000;25:648.
46.
LiCCaiCTaoY, et al. Variation and evolution of the whole chloroplast genomes of Fragaria spp. (Rosaceae). Front Plant Sci. 2021;12:754209. doi:10.3389/fpls.2021.754209
47.
DoHDKKimCChaseMWKimJH. Implications of plastome evolution in the true lilies (monocot order Liliales). Mol Phylogenet Evol. 2020;148:106818.
48.
LamVKSoto GomezMGrahamSW.The highly reduced plastome of mycoheterotrophic Sciaphila (Triuridaceae) is colinear with its green relatives and is under strong purifying selection. Genome Biol Evol. 2015;7:2220-2236.
49.
GruzdevEVMardanovAVBeletskyAV, et al. The complete chloroplast genome of parasitic flowering plant Monotropa hypopitys: extensive gene losses and size reduction. Mitochondrial DNA B. 2016;1:212-213.
50.
RoquetCCoissac CruaudC, et al. Understanding the evolution of holoparasitic plants: the complete plastid genome of the holoparasite Cytinus hypocistis (Cytinaceae). Ann Bot. 2016;118:885-896.
51.
WheelerGLDormanHEBuchananAChallagundlaLWallaceLE.A review of the prevalence, utility, and caveats of using chloroplast simple sequence repeats for studies of plant biology. Appl Plant Sci. 2014;2(12):apps.1400059.
52.
DeguillouxM-FPemongeM-HPetitRJ.Use of chloroplast microsatellites to differentiate oak populations. Ann For Sci. 2004;61:825-830.
53.
WestwoodJHYoderJITimkoMPdePamphilisCW.The evolution of parasitism in plants. Trends Plant Sci. 2010;15:227-235.
54.
Cauz-SantosLAda CostaZPCallotC, et al. A repertory of rearrangements and the loss of an inverted repeat region in Passiflora chloroplast genomes. Genome Biol Evol. 2020;12:1841-1857.
55.
UedaMFujimotoMArimuraS, et al. Loss of the rpl32 gene from the chloroplast genome and subsequent acquisition of a preexisting transit peptide within the nuclear gene in Populus. Gene. 2007;402:51-56.
56.
AlqahtaniAAJansenRK.The evolutionary fate of rpl32 and rps16 losses in the Euphorbia schimperi (Euphorbiaceae) plastome. Sci Rep. 2021;11:7466.
57.
MohantaTKMishraAKKhanA, et al. Gene loss and evolution of the Plastome. Genes. 2020;11:1133.
58.
XiongA-SPengR-HZhuangJ, et al. Gene duplication, transfer, and evolution in the chloroplast genome. Biotechnol Adv. 2009;27:340-347.
59.
TillerNBockR.The translational apparatus of plastids and its role in plant development. Mol Plant. 2014;7:1105-1120.
60.
DobrogojskiJAdamiecMLucińskiR.The chloroplast genome: a review. Acta Physiol Plant . 2020;42:98.
61.
MageeAMAspinallSRiceDW, et al. Localized hypermutation and associated gene losses in legume chloroplast genomes. Genome Res. 2010;20:1700-1710.
62.
DoHDKKimJ-H.A dynamic tandem repeat in monocotyledons inferred from a comparative analysis of chloroplast genomes in Melanthiaceae. Front Plant Sci. 2017;8:693. doi:10.3389/fpls.2017.00693
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.