
Abstract
Keywords
Introduction
Activation of the TLR4-myeloid differentiation 2 (MD-2) complex, an essential component of the mammalian innate immune system, by LPS results in a systemic inflammatory response. One of the major concerns in TLR4 signaling is the ability of the active LPS–MD-2·TLR4 complex to overstimulate the innate immune system causing life-threatening health conditions such as sepsis syndrome and septic shock.1,2 Down-regulation of TLR4 signaling was shown to be beneficial for treatment of many chronic and acute inflammatory diseases such as asthma,
3
arthritis,
4
influenza
5
and cancer.
6
The ‘endotoxic principle’ of LPS resides in a glycophospholipid lipid A, which can be recognized and bound by the MD-2·TLR4 complex.
7
The chemical structure of lipid A is based on the β(1→6)-linked 1-,4′-bisphosphorylated diglucosamine backbone, which is multiply acylated by (
Conformationally confined tetraacylated lipid A mimetics (LAM) based on the βGlcN(1↔1)αGlcN scaffold were designed and synthesized to investigate the molecular basis of the disruption of MD-2·TLR4-mediated inflammatory signaling. 9 The idea of developing conformationally restricted lipid A mimetics was driven by the pioneering studies of the group of Seydel and Brandenburg, that disclosed a correlation between the endotoxic activity and the shape and volume of the hydrophobic region of lipid A enclosed in the lipid matrix, which was dependent on the inclination of the backbone with respect to the acyl chains.10–13 Furthermore, outstanding synthetic research by the groups of Fukase and Kusumoto also suggested the existence of a relationship between the molecular shape of a single lipid A molecule and its biological activity.14–16 In the inspiring seminal work of Zähringer and Grzesiek, the three-dimensional (3D) conformation of a monomeric LPS molecule was deciphered by intricate NMR experiments revealing a correlation between the relative orientation of the GlcN units of the βGlcN(1→6)GlcN lipid A backbone in differently acylated LPS structures. 17
In the βGlcN(1↔1)αGlcN LAMs the inherently flexible three-bond β(1→6) glycosidic linkage of the carbohydrate backbone of native lipid A is displaced by a rigid two bond βα(1↔1) glycosidic linkage (Figure 1). Thus, βGlcN(1↔1)αGlcN-based LAMs represent conformationally restricted counterparts of the biosynthetic precursor of (Top) Chemical structure of βGlcN(1↔1)αGlcN-based LAM DA193 having a rigid two-bond linked β,α(1↔1) diglucosamine backbone in comparison with the flexible three-bond linked β(1→6) diglucosamine backbone of native lipid A and lipid IVa. (Bottom) Antagonist DA193 (stick model, yellow) in the binding pocket of h- and mMD-2. (A) Superimposition of DA193hMD-2 (obtained by molecular dynamic simulations) and 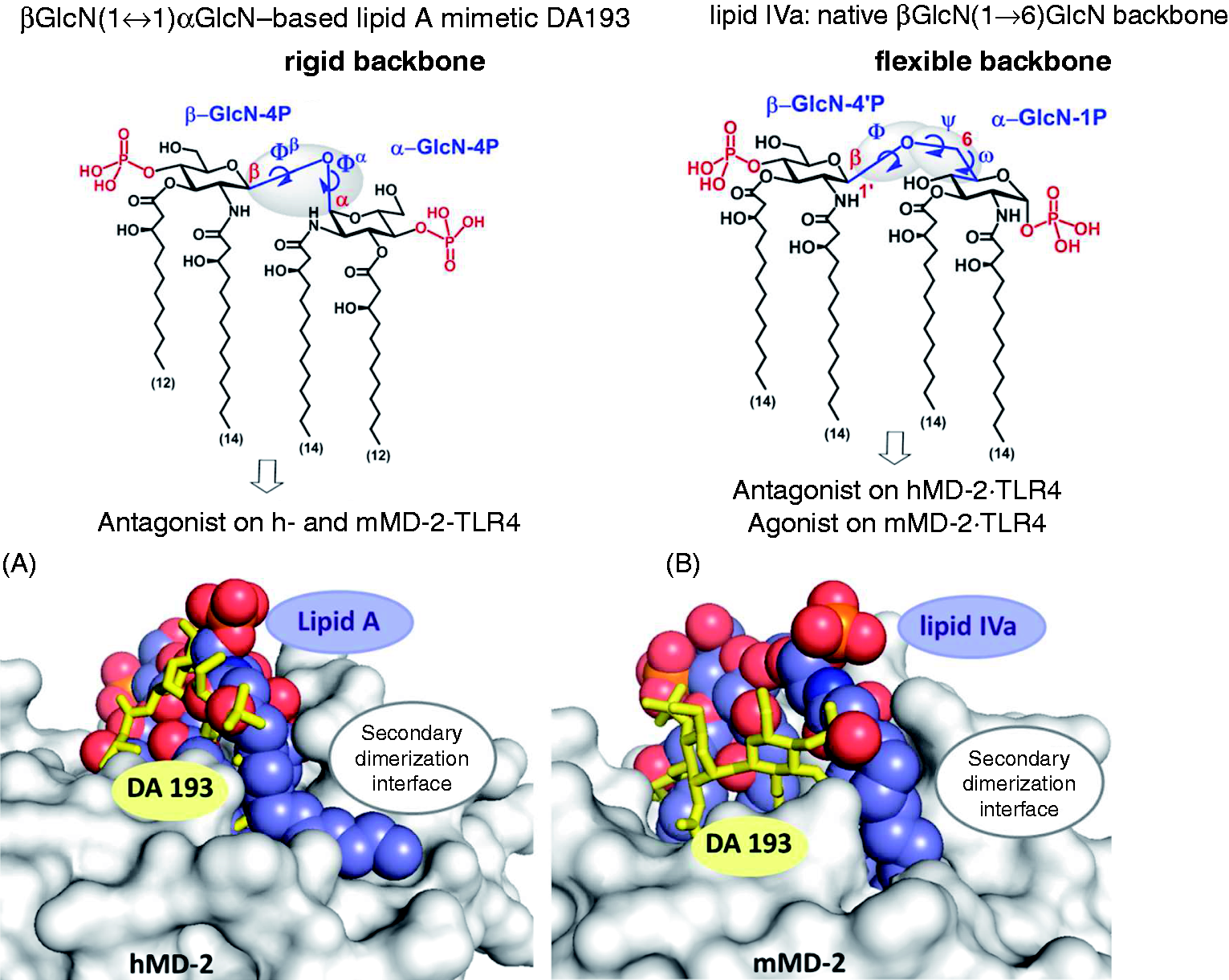
To further investigate the antagonistic properties of tetraacylated βGlcN(1↔1)αGlcN-based lipid A mimetics in human cells, we examined the ability of six variably acylated βGlcN(1↔1)αGlcN LAMs to inhibit LPS-induced pro-inflammatory responses in human macrophage-like cell line (THP-1), in human epithelial cells and in human immune cells [dendritic cells (DCs)].
One of the most striking and still not clarified aspects disclosed in the co-crystal structures of MD-2·TLR4 ligand complexes is that the orientation of the agonistic ligands (such as hexaacylated Orientations of the lipid A ligands within the binding pocket of MD-2. (A) Schematic representation of the orientation of the agonistic ligands resolved in the co-crystal structures: 
Materials and methods
Biological assays
Reagents
THP-1 cells were a kind gift of R. De Vos (Roche Ghent); the cell culture medium RPMI 1640 (Life Technologies, Carlsbad, CA, USA) was supplemented with 2 mM
Assay in THP-1 cells
THP-1 cells [human acute monocytic leukemia cell line induced for monocytic differentiation with 12-
Assay in DCs
Cell preparation and stimulation
PBMCs were isolated from heparinized whole blood (buffy coats) of healthy donors purchased from the Red Cross in Austria by standard density gradient centrifugation with Ficoll-Paque (Pharmacia Biotech, Piscataway, NJ, USA). Subsequently, monocytes and T cells were separated by magnetic sorting using the MACS technique (Miltenyi Biotec, Cologne, Germany) as previously described.
25
Monocytes were enriched using the biotinylated CD14 mAbs VIM13 and MEM18 (purity >95%). Purified T cells were obtained through negative depletion of CD11b, CD14, CD16, CD19, CD33 and MHC class II-positive cells with the respective mAbs. DCs were generated from CD14+ monocytes cultured in the presence of GM-CSF (50 ng/ml) and IL-4 (100 U/ml) for 6 d. Maturation of DCs was induced by adding 10 ng/ml
Immunofluorescence analysis
For membrane staining, cells (5 × 105) were incubated for 30 min at 4℃ with unlabeled mAbs at a concentration of 20 µg/ml. Staining of DCs was performed in the presence of human IgG Abs (20 mg/ml; Beriglobin; Aventis Behring Marburg, Germany). After washing cells twice with ice-cold PBS containing 1% BSA, binding of the primary mAb was visualized by the use of Oregon Green-conjugated goat anti-mouse Ab from Molecular Probes (Eugene, OR, USA). Cells were then washed three times with PBS/BSA. Membrane fluorescence was analyzed on a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA, USA) supported by CellQuest-Pro software (BD Biosciences). The exclusion of dead cells was performed by the addition of propidium iodide.
Determination of cytokine production
DCs were treated as indicated, and after 24 h the supernatants were harvested and analyzed by Luminex (Austin, TX, USA) testing for the presence of TNF-α, IL-10, IL-12p70 and IL-6 using specific matched-pair Abs and recombinant cytokines as standards (eBioscience, San Diego, CA, USA) as described. 26 Cytokine measurements were performed in duplicates using the Luminex System 100. Results are representative of three independent experiments (mean values of triplicate examinations ± SD are presented in Figures 4 and 5).
Assay in epithelial cells
Beas-2 b (an Ad12SV40-transformed human bronchioepithelial cell line) or Calu-3 cells (a human lung epithelial cell line; both from the ATCC) were seeded in 96-well plates at 100,000 cells/well in 100 μl of complete medium [RPMI1640 (PAA Laboratories, Pasching, Austria), 1% PS (PAA Laboratories, Pasching, Austria), 10% FCS (Biochrom, Berlin, Germany)]. On the next day, the cells were washed once with complete medium. Before stimulation with 10 ng/ml
NMR spectroscopy
Two sets of experiments were performed using DMSO-d6 or D2O/deuterated SDS micelles as solvent. NMR experiments were recorded at 298 K on a Bruker AV500 spectrometer (Bruker, Billerica, MA, USA). Spectra were obtained with standard sequences from the TOPSPIN software package (Bruker). For the DMSO experiments, a ∼ 2 mM concentration for the βGlcN(1↔1)αGlcN-LAMs was employed. For experiments in the presence of deuterated SDS micelles, 23.80 μl of a stock solution of DA257 (4.2 mM in DMSO-d6) was mixed with 45.5 μl of SDS and further diluted with D2O to give a final concentration of 0.5 ml of the ligand in the presence of SDS (20 mM). The components were mixed up by vortexing. The Bruker pulse sequence
Molecular dynamics simulation
Model building is described in the Supporting Information.
Simulation setup
All simulations were performed with the program NAMDv2.9. 27 The Particle Mesh Ewald method was used for long-range electrostatics within a relative tolerance of 1.10−6. 28 A cut-off distance of 1.2 nm was applied to real-space Ewald interactions and for the van der Waals interactions, with a smooth switching function applied between 1.0 and 1.2 nm. Multiple time steps were used with time steps of 2 fs for bonded interactions, 2 fs for short-range non-bonded interactions and 4 fs for the full electrostatics evaluation using the r-RESPA method. All production runs were performed at constant temperature and pressure, with reference values of 298 K and 1 atm, using the Nose-Hoover and Langevin piston methods with damping coefficients of 1 ps–1.29,30 The SHAKE algorithm was applied to constrain bond lengths to all hydrogen atoms. 31 After an initial thermalization, all simulations were run for 11 ns, discarding the first ns.
Analysis of molecular dynamics simulations
Hydrogen bonds of the ligands with the solvent and the protein were identified by geometric criteria: a donor–acceptor pair is considered to be hydrogen bonded if the donor–acceptor distance is < 0.35 nm and the donor–hydrogen–acceptor angle is >150°. Salt bridges between the negatively charged phosphates of the lipids and the positively charged residues of MD-2 were determined using a cut-off of 0.7 nm between the central phosphorous atoms and the Cz of arginine or the Nz of lysine, respectively. Free energy difference (ΔGbind) calculations were computed using the linear interaction energy (LIE) method, 32 with the empirical parameters α = 0.18 and β = 0.09. 33 As NOE intensities for slowly tumbling molecules are proportional to the inverse distance to the third power, average distances from the simulations were calculated as <r−3>−1/3, with angular brackets indicating an ensemble average.
Results
Antiendotoxic potential of βGlcN(1↔1)αGlcN-based lipid A mimetics in human macrophage-like cell line THP-1
Six variably acylated βGlcN(1↔1)αGlcN-based lipid A mimetics were examined for the inhibition of expression of TNF-α in the human monocytic cell line THP-1 that was differentiated into macrophages by TPA treatment (Figure 3). Though THP-1 cells are derived from the blood sample of a patient with acute monocytic leukemia, this cell line resembles primary monocytes and macrophages from healthy donors.
24
Inhibition of the expression of TNF-α by βGlcN(1↔1)αGlcN-LAMs in THP-1 cells stimulated with 100 ng/ml 
Macrophage-like THP-1 cells express MD-2, mCD14 (which is required for the transfer of monomeric LPS to the MD-2·TLR4 complex) and a variety of cell surface receptors, including TLR4.
34
The cells were stimulated by
Anti-endotoxic activity of βGlcN(1↔1)αGlcN-based lipid A mimetics in human DCs
DCs, which are professional pathogen-decoding APCs, reside in peripheral tissues as immature APCs that can be activated by PAMPs, 35 and, in particular, by LPS. 36 LPS-stimulated DCs mature to the competent T cells by translocating MHC–peptide complexes to the cell surface and up-regulating co-stimulatory cell surface receptors. Besides, activated DCs produce a number of cytokines engaged in elimination of infection and modulation of the T-cell responses. 37
The ability of selected βGlcN(1↔1)αGlcN LAMs, which revealed the highest potency in inhibiting the activation of NF-κB in the HEK-blue cells,
9
to interfere with LPS-induced NF-κB and IFN regulatory factor (IRF) inflammatory pathways by inhibiting the up-regulation of specific surface markers on DCs was initially assessed. To test the impact of βGlcN(1↔1)αGlcN LAMs on DC maturation, immature, monocyte-derived DCs were treated with LPS with or without the addition of four variably acylated βGlcN(1↔1)αGlcN LAMs. DCs treated with LPS acquired a characteristic morphologic phenotype and displayed specific markers of mature DCs when analyzed by flow cytometry (Figure 4). Treatment of DCs stimulated by Inhibition of expression of maturation markers induced by 
The capacity of the synthetic TLR4 antagonists to impede the LPS-induced expression of pro-inflammatory cytokines in human DCs was assessed by incubation of immature monocytes-derived DCs with LPS in the presence of βGlcN(1↔1)αGlcN LAMs. The βGlcN(1↔1)αGlcN LAMs DA193, DA254, DA256 and DA257 were examined for efficacy in prohibiting the induction of cytokines that are normally released by DCs during acute infections or upon Inhibition of expression of cytokines by βGlcN(1↔1)αGlcN-based LAMs in monocyte-derived DCs induced by 
Evaluation of antagonistic activity of βGlcN(1↔1)αGlcN-based lipid A mimetics in human epithelial cells
We assessed the antagonistic activity of the βGlcN(1↔1)αGlcN LAMs in two different human lung epithelial cell lines, BEAS-2B and Calu-3, which express TLR4 and MD-2, but do not express mCD14. With respect to the inhibition of IL-8 release, both cell lines behaved nearly identically (Figure 6A, B) revealing that DA193 and DA256 were the most potent antagonists, with a concentration of 100 ng/ml sufficient to reduce cytokine release to almost baseline levels (> 90% inhibition). DA253 and DA254 were somewhat less effective, providing > 80% inhibition at 100 ng/ml antagonist. At the same concentration, shorter-chain DA255 reduced cytokine release only by 50–60%. However, all compounds except for DA257 completely suppressed activation when used at a concentration of 1000 ng/ml. A nearly identical pattern of the antagonistic capacity could be seen for the inhibition of IL-6 release (data not shown).
Inhibition of production of IL-8 by βGlcN(1↔1)αGlcN LAMs in human epithelial cell lines (A) BEAS-2b and (B) Calu-3 stimulated by addition of 10 ng/ml 
Assessment of the conformation about β,α(1↔1) glycosidic linkage by NMR
The potent antagonistic activity of the βGlcN(1↔1)αGlcN LAMs was attributed to a specific molecular shape of the conformationally confined βGlcN(1↔1)αGlcN backbone. Therefore, the experimental assessment of the conformation about βα−(1↔1) glycosidic linkage by NOESY experiments of variably acylated synthetic glycolipids (DA193, DA256 and DA257) was performed.
39
Given the poor solubility of glycolipids in water, the NMR data were acquired first in DMSO-d6 followed by D2O–SDS micelles, taking advantage of the fact that the basic conformational features of glycolipids in different solvent media are preserved.40,41 The analysis of the vicinal coupling constants indicated that both Superposition of 1H-NMR spectrum of DA193 in DMSO-d6 (red) with TOCSY (yellow) and NOESY (blue) spectra. The computed proton–proton distances for the diglucosamine scaffold of DA193 are shown.
As DMSO is known to disrupt hydrophobic interactions between lipid chains that could influence the overall conformation of the molecule, we analyzed the conformational behavior of lipid A mimetics in D2O supplemented by SDS micelles. Given the similarity of the NMR spectra of variably acylated βGlcN(1↔1)αGlcN LAMs in DMSO, the analysis was carried out for the shorter-chain DA257, taking advantage of its better solubility in water/SDS. Notably, the resolution of the 1H-NMR spectrum of DA257 in the SDS micelle environment was significantly improved with respect to that in DMSO (online Supplementary Figure S4). Acquisition of the NOESY spectrum allowed for assignment of the strong cross peak H1α/H1β and assessment of the conformation of glycosidic linkage, which was similar to the value obtained in DMSO. The cross peaks were already negative, indicating the large size of the aggregates in solution and, again, the predominant
Molecular dynamics simulation of antagonist DA193 in the binding pocket of hMD-2
Molecular dynamics simulations of DA193, the most potent hMD-2·TLR4 antagonist in the series of βGlcN(1↔1)αGlcN LAMs, were performed to gain a deeper insight into the molecular basis of the ligand recognition by hMD-2. Two possible orientations (rotation by 180°) of DA193 in the binding pocket of hMD-2 were simulated and compared in geometric characteristics and binding affinities to the modeled MD-2-bound native ligands
The calculated average values of the Φα/Φβ torsions and, consequently, H1α–H1β and H2β–H5α distances for the protein-bound DA193, as well as for DA193, placed at the octane–water interface were in excellent agreement with the experimentally determined (NOESY) upper bounds, whereas the maximum violation amounted to 0.018 nm (Figure 8A). Overall, the calculated H1α–H1β and H2β–H5α distances and the corresponding torsions only insignificantly deviated from the experimentally observed values. Moreover, the 3D arrangement of the diglucosamine backbone of DA193 minimized in the protein-bound state only slightly deviated from the conformation of β,α-trehalose found in the x-ray structure,
45
which can be explained by the rigidity of βα(1↔1)glycosidic linkage and the independence of the conformation on the nature and number of substituents (online Supplementary Figure S5).
(A) Conformation of the βGlcN(1↔1)αGlcN backbone of DA193 simulated in the binding pocket of hMD-2 and at an octane–water interface compared with the conformation experimentally obtained from the NOESY NMR experiments of the ligands in solutions. Images were generated with PyMol. Averages of MD simulations were calculated as <r-3>–1/3 and error estimates were obtained from block averaging. NMR distances are considered as upper bounds such that the length longer than the experimental value is a violation, while any value that is lower than the experimental value is allowed. (B) Superimposition of antagonistic lipid IVahMD-2 (light orange, PDB code: 2E59) with (A) simulated DA193hMD-2 (blue) in pose A and (B) with simulated DA193hMD-2 in pose B; hMD-2 is not shown for clarity.
The distance between 4 and 4′ phosphate groups in DA193 only marginally differed from the P1–P4′ distances in the native ligands found in the co-crystal structures (online Supplementary Table S1). Apparently, the plasticity of the phosphate groups on one side and the rigidity of β,α−(1↔1) glycosidic linkage which keeps two (1↔1)-connected GlcN rings in a nearly co-planar arrangement on the other side readily compensate for the shorter two-bond linkage between the GlcN rings in DA193 compared with the three-bond (1→6) glycosidic linkage in native lipid A and lipid IVa (Figure 8B).
Average occurrence of hydrogen bonds and salt bridges for lipid A, lipid IVa and DA193 in complex with hMD-2, together with the atom positional root mean square displacement (RMSD) of Phe126. a

Error estimates are calculated from block averages.
Average total number of hydrogen bonds, including the protein and the solvent. In pure water or octane–water environment, the number of hydrogen bonds is, on average, 14.5 (lipid A); 13.6 (lipid IVa); 12.8 (DA193) nm.
Atom-positional RMSD of Phe126 at the end of the simulations with respect to the initial MD-2 structure calculated after a rotational fit on the Cα atoms of the MD-2 backbone. All simulations were started from the agonistic MD-2 structure (3FXI). The Phe126 remained relatively close to its original position (small RMSD) in the simulations of lipid AhMD-2 in pose A, while the crystallographically not-observed binding poses (lipid A in pose B and lipid IVa in pose A) induced a larger conformational change of Phe126. Larger deviations are observed when hMD-2 antagonist (lipid IVa or DA193) is placed in an agonistic protein structure (3FXI). Thus, the dynamic behavior of Phe126 was in line with the antagonistic role of DA193.

Electrostatic interactions at the rim of the binding pocket of hMD-2. (A) 4,4′-phosphate groups (in red) of modeled DA193 (Pose B) are involved in intense ionic contacts with Lys122, Lys125, Arg90 and Lys58; moderate interactions are detected with Arg96. (B) 1, 4′-Phosphate groups of lipid IVa (Pose B, corresponds to PDB code 2E59) establish strong ionic bridges with Lys122, Arg90 and Lys58. Images were generated with PyMol.
Relative free energy differences in kcal/mol as calculated from molecular dynamics simulations. a

Error estimates are calculated from block averages.
Free energy of binding as estimated by the LIE method: ΔGbind = αΔ<Vvdw> + βΔ<Vel>, where <Vvdw> and <Vel> are the average ligand-surrounding van der Waals and electrostatic interaction energies, Δindicates the difference between the ligand-bound and unbound simulations; α = 0.18 and β = 0.09, using exponential averages of different poses.
48
The free energy of binding is logarithmically dependent on the dissociation constant Kd: ΔG
Relative free energy difference between poses A and B, calculated using the LIE model. 33
Consistent with the expectations, DA193 did not discriminate significantly between Pose A or Pose B in hMD-2. In contrast, the control simulation of lipid AhMD-2 revealed the Pose A (corresponding to PDB code 3FXI) as a preferred orientation in hMD-2, while pose B was disfavored by 1.6 kcal/mol, which is shown by the free energy differences between the two orientations of lipid A in the binding site of hMD-2 (Table 2).
Discussion
The βGlcN(1↔1)αGlcN-based tetraacylated lipid A mimetics were designed to fit into the binding pocket of MD-2 and to compete with LPS for the same binding site on the co-receptor protein, thereby inhibiting LPS-induced receptor dimerization and the ensuing pro-inflammatory signaling. The molecular shape of the βGlcN(1↔1)αGlcN backbone of conformationally confined lipid A mimetics is supposed to imitate the spatial arrangement of the native βGlcN(1→6)GlcN backbone of hMD-2-bound antagonists Eritoran and lipid IVa disclosed in the co-crystal structures.20,21 By fixing the 3D molecular shape of the carbohydrate backbone of tetraacylated lipid A mimetics in an ‘antagonistic’ conformation through application of β,α(1↔1)-linked diglucosamine scaffold, a species-independent (human and mouse) antagonistic activity has been attained.
9
We proposed that the exposure of the 2
Since the βGlcN(1↔1)αGlcN scaffold, wherein the GlcN rings are nearly co-planar oriented, possesses a non-flexible β,α-(1↔1) glycosidic bond, the ‘flipping’of one of the GlcN moieties is not feasible. Therefore, the antagonistic nature of βGlcN(1↔1)αGlcN LAMs was supposed to be not dependent on the orientation of their carbohydrate backbone in the binding pocket of MD-2 (Figure 2C). As validated by molecular dynamics simulation, all four β-hydroxyacyl chains of DA193 in both binding orientations were fully inserted into the hydrophobic pocket of hMD-2 and the whole molecule was shifted much deeper into the binding cleft compared to the agonist
A 3D molecular shape of the β,α(1↔1)-linked diglucosamine backbone of lipid A mimetics has been ascertained by NOESY experiments of three variably acylated compounds. A
We have demonstrated that novel synthetic βGlcN(1↔1)αGlcN LAMs potently inhibit LPS-induced pro-inflammatory signaling in human DCs, the human macrophage-like THP-1 cell line and human epithelial cells. The anti-endotoxic potency of the βGlcN(1↔1)αGlcN LAMs declined, in general, with the shortening of the length of (
The 2 × C12, 2 × C14-acylated compound DA193, which inhibited the expression of the most inflammatory cytokines to the background levels at a concentration 100 ng/ml in all three human cell types tested, was highlighted as the most potent hTLR4 antagonist. Importantly, DA193 was able to inhibit equally the release of IL-6, IL-10, IL-12 and TNF-α in DCs both upon pre-incubation of the cell culture with antagonist (1 h) followed by addition of LPS and upon simultaneous treatment with LPS and DA193 (Figure 5). Powerful suppression of the release of TNF-α in the macrophage-like THP-1 cell line by three (DA193, DA256 and DA253) of six synthetic antagonists was also detected after the pretreatment of the cells with LPS (10 min) (Figure 3). Potentially, this indicates the capability of βGlcN(1↔1)αGlcN LAMs to displace competitively LPS from the binding pocket of hMD-2, which has also been verified by molecular dynamics simulations revealing 20-fold stronger affinity of DA193 to hMD-2 compared with
In conclusion, synthetic tetraacylated lipid A mimetics wherein the native βGlcN(1→6)GlcN lipid A backbone is displaced by the rigid βGlcN(1↔1)αGlcN scaffold and the labile glycosidic phosphate functionality (as in the native lipid A or in the drug candidate Eritoran) is exchanged for a stable secondary phosphate group, could serve as a basis for development of novel MD-2·TLR4 antagonists with improved anti-inflammatory activities for potential therapeutic application.
Footnotes
Funding
Conflict of interest
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.