
Abstract
Background
One major problem in applying gene expression profiles to cancer classification and prediction is that the number of features (genes) greatly surpasses the number of samples. Some studies have shown that a small collection of genes selected correctly can lead to good classification results.1–4 Therefore gene selection is crucial in molecular classification of cancer. Numerous methods of selecting informative gene groups to conduct cancer classification have been proposed. Most of the methods first ranked the genes based on certain criteria, and then selected a small set of informative genes for classification from the top-ranked genes. The most used gene ranking approaches include t-score, chi-square, information entropy-based, Relief-F, symmetric uncertainty etc.
In, 2 we used a new feature selection method for gene selection. The feature selection method was based on the α depended degree, a generalized concept of the canonical depended degree proposed in rough sets. Combining this feature selection method with decision rules-based classifiers, we achieved accurate molecular classification of cancer by a small size of genes. As pointed out in, 2 our classification methods had some advantages over other methods in such as simplicity and interpretability. Yet, there remain some essential problems to be investigated. For example, what properties does the feature selection method possess, and what will happen if we compare the feature selection method with other feature selection methods in terms of identical classifiers?
In this work, we investigated the properties of the feature selection method based on the α depended degree. We mainly studied the relationships between α value, classifier, classification accuracy and gene number. Moreover, we compared our feature selection method with other four feature selection methods often used in practice: chi-square, information gain, Relief-F and symmetric uncertainty. We chose four popular classifiers: NB (Naive Bayes), DT (Decision Tree), SVM (Support Vector Machine) and
Materials
Colon tumor dataset
The dataset contains 62 samples collected from Colon Tumor patients. 5 Among them, 40 tumor biopsies are from tumors (labeled as “negative”) and 22 normal (labeled as “positive”) biopsies are from healthy parts of the colons of the same patients. Each sample is described by 2000 genes.
Cns tumor dataset
The dataset is about patient outcome prediction for central nervous system embryonal tumor. 6 In this dataset, there are 60 observations, each of which is described by the gene expression levels of 7129 genes and a class attribute with two distinct labels—Class 1 (survivors) versus Class 0 (failures). Survivors are patients who are alive after treatment while the failures are those who succumbed to their disease. Among 60 patient samples, 21 are labeled as “Class 1” and 39 are labeled as “Class 0”.
DLBCL dataset
The dataset is about patient outcome prediction for DLBCL. 7 The total of 58 DLBCL samples are from 32 cured patients (labeled as “cured”) and 26 refractory patients (labeled as “fatal”). The gene expression profile contains 7129 genes.
Leukemia 1 dataset (ALL vs. AML)
In this dataset, 1 there are 72 observations, each of which is described by the gene expression levels of 7129 genes and a class attribute with two distinct labels—AML versus ALL.
Lung cancer dataset
The dataset is on classification of MPM (Malignant Pleural Mesothelioma) versus ADCA (Adenocarcinoma) of the lung. 8 It is composed of 181 tissue samples (31 MPM, 150 ADCA). Each sample is described by 12533 genes.
Prostate cancer dataset
The dataset is involved in prostate tumor versus normal classification. It contains 52 prostate tumor samples and 50 non-tumor prostate samples. 9 The total number of genes is 12600. Two classes are denoted as “Tumor” and “Normal”, respectively.
Breast cancer dataset
The dataset is about patient outcome prediction for breast cancer. 10 It contains 78 patient samples, 34 of which are from patients who had developed distant metastases within 5 years (labeled as “relapse”), the rest 44 samples are from patients who remained healthy from the disease after their initial diagnosis for interval of at least 5 years (labeled as “non-relapse”). The number of genes is 24481.
Leukemia 2 dataset (ALL vs. MLL vs. AML)
The dataset is about subtype prediction for leukemia. “ It contains 57 samples (20 ALL, 17 MLL and 20 AML). The number of genes is 12582.
Methods
α Depended degree-based feature selection approach
In reality, when we are faced with a collection of new data, we often want to learn about them based on pre-existing knowledge. However, most of these data cannot be precisely defined based on pre-existing knowledge, as they incorporate both definite and indefinite components. In rough sets, one
The decision table is the data form studied by rough sets. One decision table can be represented as
For the cancer classification problem, every collected set of microarray data can be represented as a decision table in the form of Table 2. In the microarray data decision table, there are
Summary of the eight gene expression datasets.

Microarray data decision table.

In rough sets, the
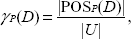
where
In some sense, γ
However, the extremely strict definition has limited its applicability. Hence, in,
2
we defined the α depended degree, a generalization form of the depended degree, and utilized the α depended degree as the basis for choosing genes. The
Comparative feature selection approaches
We compared our proposed feature selection method with the following four often used methods: chi-square, information gain, Relief-F and symmetric uncertainty.
The chi-square (χ
2
) method evaluates features individually by measuring their chi-squared statistic with respect to the classes.
15
The χ
2
value of an attribute

where
Information Gain
16
method selects the attribute with highest information gain, which measures the difference between the prior uncertainty and expected posterior uncertainty caused by attributes. The information gain by branching on an attribute

where
Relief-F method estimates the quality of features according to how well their values distinguish between examples that are near to each other. Specifically, it tries to find a good estimate of the following probability to assign as the weight for each feature
Symmetric uncertainty method compensates for information gain's bias towards features with more values. It is defined as:

where

The values of symmetric uncertainty lie between 0 and 1. A value of 1 indicates that knowing the values of either attribute completely predicts the values of the other; a value of 0 indicates that
Classification algorithms
The NB classifier is a probabilistic algorithm based on Bayes’ rule and the simple assumption that the feature values are conditionally independent given the class. Given a new sample observation, the classifier assigns it to the class with the maximum conditional probability estimate.
DT is the rule-based classifier with non-leaf nodes representing selected attributes and leaf nodes showing classification outcomes. Every path from the root to a leaf node reflects a classification rule. We use the J4.8 algorithm, which is the Java implementation of C4.5 Revision 8.
An SVM views input data as two sets of vectors in an
Data preprocess
Because chi-square, information gain, symmetric uncertainty and our feature selection methods are suitable for discrete attribute values, we need to carry out the discretization of attribute values before feature selection using these methods. We used the entropy-based discretization method, which was proposed by Fayyad et al. 19 This algorithm recursively applies an entropy minimization heuristic to discretize the continuous-valued attributes. The stop of the recursive step for this algorithm depends on the MDL (Minimum Description Length) principle.
Feature selection and classification
We ranked the genes in a descendent order of their α depended degree, and then used the top 100, 50, 20, 10, 5, 2 and 1 genes for classification with the four classifiers, respectively. In addition, we observed the classification results with the seven different α values: 1, 0.95, 0.9, 0.85, 0.8, 0.75 and 0.7. Moreover, we used the top 100, 50, 20, 10, 5, 2 and 1 genes ranked by the other four feature selection methods for classification with the four classifiers, respectively. Considering that the sample size in every dataset was relatively small, we used LOOCV (Leave-One-Out Cross-Validation) method to test the classification accuracy.
We implemented the data preprocess, feature selection and classification algorithms mainly in the Weka package. 20
Results and Analysis
Classification results using our feature selection method
Table 3 shows the classification results based on the α depended degree in the Colon Tumor dataset. The classification results in the other datasets based on the α depended degree were provided in the supplementary materials (1).
Classification accuracy (%) in the Colon tumor dataset based on the α depended degree.

The maximum numbers in each row are highlighted in boldface, indicating the highest classification accuracy achieved by among the different classifiers under the identical α value and gene number.
Comparison of classification performance for different classifiers
Table 3 shows that there are in total 12, 19, 11 and 20 best classification cases for NB, DT, SVM and
Number of best classification cases among the different classifiers.

The maximum numbers in each row are highlighted in boldface.

Best classification accuracy of each classifier.
In addition, we considered the average classification performance. Table 5 shows the respective average classification accuracy of the four classifiers under different α values in the Colon Tumor dataset. The results revealed that the
Average classification accuracy (%) for the different classifiers and α values in the Colon tumor dataset.

The maximum numbers in each row are highlighted in boldface.
Number of the best average classification performances achieved by each classifier under various α values for each dataset.

The maximum numbers in each row are highlighted in boldface.

Average classification accuracy of each classifier.
The optimum gene size for classification depends on different classification algorithms. We found DT generally used fewer genes to reach the best accuracy compared with the other classification algorithms. This is one advantage of DT learning algorithm in that DT is a rule-based classifier and fewer genes will induce simpler classification rules, which in turn facilitate the interpretability of DT models.
Depended degree vs. α depended degree
The depended degree was commonly applied in feature selection in rough sets-based machine learning and data mining. However, our recent studies have revealed that for the microarray-based cancer classification problem, the application of the depended degree was severely limited because of its overly rigor definition. In contrast, its generalized form-α depended degree, had essentially improved utility.
2
To explore how the classification quality was improved by using the α depended degree relative to the depended degree, we compared the classification results obtained under different α values while based on the identical classifiers. Figure 3 lists the average classification accuracies for different α values under the four different classifiers in the Colon Tumor dataset. The results shows that when NB was used for classification, the average classification accuracy in the case of the depended degree (α = 1) was only slightly better than the case of α = 0.95 and worse than all the other cases; when DT was used for classification, the average classification accuracy with the depended degree was the poorest; When SVM or

Average classification accuracy for different α values.
Further, we compared the best classification situations obtained under different α values. As shown in Table 7 and Figure 4, for the Colon Tumor dataset, in the cases of NB and DT, the best results were obtained when α = 0.85 and α = 0.9, respectively, although in the cases of SVM and
Best classification accuracy (%) for the different classifiers and α values in the Colon tumor dataset.

The maximum numbers in each column are highlighted in boldface.

Best classification accuracy for different α values.
All together, the α depended degree is a more effective feature selection method compared to the conventional depended degree.
Interrelation between classification accuracy and α value
In the previous studies,
2
we intuitively felt that the α value had some connections with inherent characters of related datasets. If the best classification accuracy was achieved only under relatively low α values, the dataset might be involved in relatively difficult classification and high classification accuracy would be hard to achieve. To prove this conjecture, we first detected the highest classification accuracies and their corresponding α values for each classifier, and calculated the averages of the accuracies and the averages of the α values under the four classifiers. For example, from Table 3, we knew that in the Colon Tumor dataset, NB had the highest accuracy of 88.71% accompanying with α = 0.85; DT had the highest accuracy of 91.93% accompanying with α = 0.9; SVM had the highest accuracy of 88.71% accompanying with α = 1, 0.9 and 0.7;

and the average of the α values as follows:

We call this kind of average accuracies the
In addition, we calculated the average classification accuracy for each α-classifier pair, and found the best average accuracy and its corresponding α value for each classifier. Likewise, we calculated their averages under the four classifiers. For example, from Table 5, we knew that in the Colon Tumor dataset, α-NB had the best average accuracy of 83.64% with α = 0.8 and 0.85; α-DT had the best average accuracy of 83.87% with α = 0.8; α-SVM had the best average accuracy of 84.33% with α = 0.85; α-

and the average of the α values as follows:

We call this kind of average accuracies the
Average highest and best average classification accuracy (%).

The relatively bigger AHA, ABAA, α values and their corresponding datasets are highlighted in boldface, while the relatively smaller ones are highlighted in italic.
Figure 5 and Figure 6 reflect the alteration tendencies of AHA and ABAA along with the variation of α value, respectively. In general, AHA and ABAA increase with the growth of α value except for a few exceptions. Therefore, to a certain degree, the α depended degree can reflect the classification difficultness for a certain dataset, indicating the inherent biology of specific cancers. Indeed, the classification of Leukemia 1, Lung Cancer, Prostate Cancer and Leukemia 2 has been commonly recognized as relatively easy while the classification of Breast Cancer and DLBCL relatively difficult. Our results lend support to these findings.

relationship between AHA and α.

relationship between ABAA and α.
To further investigate the relationship between classification difficultness and α, we used the co-ordinates graph to show under different α values, the average and best classification results using every classifier. Figure 7 and Figure 8 show the results for the Colon Tumor dataset. From both figures, we inferred that in the dataset, the α values of between 0.8 and 0.9 would result to the best classification accuracy generally. We stated such α value the

Average accuracy under each α value in Colon tumor.

Best accuracy under each α value in Colon tumor.
Table 9 presents the overall average and best classification performance, as well as the optimum α value for every dataset in terms of all of the four classifiers. Clearly, those datasets with higher classification accuracies have the bigger optimum α values in general. For example, the Leukemia 1, Lung Cancer, Prostate Cancer and Leukemia 2 datasets with relatively higher average and best classification accuracies have obviously larger optimum α values than the other datasets. In contrast, the DLBCL and Breast Cancer datasets have worse classification results, and smaller optimum α values. The conditions of Colon and CNS Tumor datasets are just lying between. These results again proved our conjecture that the α value was connected with the inherent classification property that a dataset possesses. Therefore, to achieve better classification of different datasets, the flexible tuning of α parameter is necessary. It is just the main advantage of the α depended degree over the depended degree.
Overall average and best classification accuracy (%) and optimum α value.

The relatively higher average accuracies, best accuracies, optimum α values and their corresponding datasets are highlighted in boldface, while the relatively lower ones are highlighted in italic.
Classification results based on other feature selection methods
Table 10 lists the classification results based on Chi (chi-square), Info (information gain), RF (Relief-F) and SU (symmetric uncertainty) in the Colon Tumor dataset. The classification results in the other datasets based on the same feature selection methods were provided in the supplementary materials (5). To verify the aforementioned inherent classification difficultness of related datasets, we calculated the highest and average of all of the classification results obtained by the different feature selection methods except the α depended degree, gene numbers and classifiers for each dataset. The results were listed in Table 11, indicating again that the Leukemia 1, Lung Cancer, Prostate Cancer and Leukemia 2 datasets can be classified with relatively high accuracy; the DLBCL and Breast Cancer datasets can be classified with relatively low accuracy; the Colon and CNS Tumor datasets can be classified with intermediate accuracy.
Classification results in the Colon tumor dataset based on the other feature selection methods.

The best classification accuracies on each combination of feature selection methods and classifiers are indicated by boldface.
Highest and average classification accuracy (%) for each dataset.

The relatively higher highest accuracies, average accuracies and their corresponding datasets are highlighted in boldface, while the relatively lower ones are hightlighted in italic.
Comparison between α depended degree and other feature selection methods
We compared the α depended degree with the other feature selection methods in the average and best classification accuracy. Table 12 lists the average classification accuracies resulted from different feature selection methods in the Colon Tumor dataset. When α = 0.85 and α = 0.80, we obtained 84.33% and 83.81% accuracy (shown in boldface), respectively. Both results exceed the results derived from Chi, Info, RF and SU.
Comparison of average classification accuracy in Colon tumor dataset.

The two largest average values are highlighted in boldface.
Table 13 lists the best classification accuracy obtained by different feature selection methods in the Colon Tumor dataset. For the classifier NB, the maximum best classification accuracy of 88.71% was obtained under chi and α = 0.85; for DT, the maximum was obtained under SU and α = 0.9; for SVM, the maximum was achieved under α = 1, 0.9 and 0.7; for
Comparison of best classification accuracy in Colon tumor dataset.

The maximums of each column are shown in boldface, indicating the highest best classification accuracies obtained among the different feature selection methods using the identical classifiers.
Figure 9 and Figure 10 contrast the average and best classification accuracies in all of the eight datasets for different feature selection methods. In the average accuracy, the α depended degree attained the best results in four datasets; in the best accuracy, the α depended degree attained the best results in six datasets. Taken together, the classification performance with the α depended degree are superior to or at least match that with the other four popular feature selection approaches.

Contrast in average accuracy for different feature selection methods.

Contrast in best accuracy for different feature selection methods.
Discussion and conclusions
Because of the severe imbalance between feature numbers and instance numbers in microarray-based gene expression profiles, feature selection is essentially crucial in addressing the problem of molecular classification and identifying important biomarkers of cancer. To better molecularly classify cancers and detect significant marker genes, developing flexible and robust feature selection methods are of extreme importance. However, the conventional rough sets based feature selection method, the depended degree of attributes, was deficient in flexibility and robustness. Some indeed important genes may be missed just as their exceptional expression in a small number of samples if the depended degree criterion is used for gene selection. In contrast, we can avoid this kind of situations by the utility of the α depended degree criterion, which shows strong robustness by the flexible tuning of the α value. The α depended degree has been proven to be more efficient than the depended degree in gene selection through a series of classification experiments. Moreover, the α depended degree was comparable with the other established feature selection standards: chi-square, information gain, Relief-F and symmetric uncertainty, which was also demonstrated by the classification experiments. It should be noted that the classification results exhibited in this work might be biased towards higher estimates since the feature selections were ahead of LOOCV. However, the comparisons were generally just because all of the classification results were obtained based on the same procedures.
An interesting finding in the present study was that the α depended degree could reflect the inherent classification difficultness of one microarray dataset. Generally speaking, when the α depended degree was used for gene selection, if we could achieve the comparatively good classification accuracy in some cancerous microarray dataset, the corresponding α value would be relatively high, regardless of what classifier being used; otherwise, it would be relatively low. Moreover, once some dataset has been identified as difficultly-classified or easily-classified through the α depended degree, the dataset would be equally difficultly-classified or easily-classified using other gene selection methods, irrespective of classifiers. Therefore, if we want to gauge the difficultness of the cancer-related classification based on a new microarray dataset, the α depended degree can be used for addressing the problem. In fact, if excluding the quality factor of a cancerous microarray dataset, the classification difficultness of the dataset might reflect the essential biological properties of the relevant cancer.
The size of the selected gene subset by which a good classification is achieved is also an important factor in assessing the quality of a feature selection approach. In general, the accurate classification with a small size of genes is the better classification than that with a large number of genes. Our experiments did not exhibit substantial differences in the optimum gene numbers concerned with every feature selection method, partly because finding the optimum gene sizes need more delicate feature selection strategies instead of simply selecting a few top-ranked genes. One of our future work is to develop more favorable gene selection methods by merging the α depended degree based feature ranking with some heuristic strategies.
Disclosures
This manuscript has been read and approved by all authors. This paper is unique and is not under consideration by any other publication and has not been published elsewhere. The authors report no conflicts of interest.