
Abstract
Keywords
Introduction
Improvements in cancer classification have been of great importance in cancer treatment. Conventional diagnostic methods are based on subjective evaluation of the morphological appearance of the tissue sample, which requires a visible phenotype and a trained pathologist to interpret the view. 1 It is difficult for those approaches to distinguish tumours with similar histopathological appearance (phenotype) but different clinical course and response to therapy. Since the advent of microarray technology, researchers have begun to use expression array analysis as a quantitative phenotyping tool. The potential advantage of using arrays for phenotyping is that they provide a simultaneous quantitative measure of thousands of parameters (gene expression levels) some of which are likely to have disease relevance. Due to the ability to quantify a large number of parameters, the use of expression array in diagnosing, promises both more accurate class prediction and the identification of subclasses that could not be defined by traditional methods. Even though this technology is promising in disease diagnosis, there are many huddles that the researcher should over come in order to achieve such goals. Since extraction of mRNA from a single cell is extremely difficult task, researchers are forced to pool tissues that seem to share the same fate or the same functions to obtain the adequate quantity of mRNA. This may imply that the expression levels calculated are the means of all the cells in the pool. Another issue is with the genetic variability of two individuals that will affect the expression of genes. The accumulation of noise to affect the outcome, at various points of experiment is yet another issue. Finally, the samples collected are small in numbers that results in high dimensional data with very large (thousands to tens of thousands) number of genes. The most fundamental problem that needs to solve the above mentioned caveats of the technology is to identify genes whose expression patterns either characterize a particular cell state or predict a certain forthcoming cell state. The first step in solving this problem is the development of tools for classifying samples according to their gene expression. Various clustering, classification and predicting techniques have been used to analyze and understand the gene expression data resulted from DNA microarray. Some recent applications include: molecular classification of acute leukaemia, 1 classification of human cancer cell lines, 2 Support Vector Machine (SVM) classification of cancer samples. 3
A challenge in predicting diagnostic categories using microarray data is that the number of genes is usually significantly greater than the number of tissue samples available, and only a subset of the genes is relevant in distinguishing different classes. Selection of relevant genes for classification is known as feature selection. This has three main applications: first, the classification accuracy is often improved using a subset instead of the entire set of genes: second, a small set of relevant genes is convenient for developing diagnostic tests; and third, these genes may lead to biologically interesting insights that are characteristics of the classes of interest. There have been many reports that address the classification and feature-selection problems. 4 However, many of these methods are tailored towards binary classification in which there are only two classes. While majority of the cancer phenotypes have more than two subtypes, which leads us to a multi-class prediction scenario.
Multiple class prediction is more difficult than binary prediction because the classification algorithm has to consider a greater number of separation boundaries or relations..5,6 In binary classification, an algorithm can make a decision boundary for only one of the classes; the other class is simply the complement. In multi-class classification problems, each class has to be defined explicitly. A multi-class problem can be decomposed into a set of binary problems and then combined to make a final multi-class prediction. The general term used for such procedures are called Ensemble methods.
The basic idea behind combining binary classifiers is to decompose the multiclass problem into a set of easier and more accessible binary problems. The main advantage in this divide-and-conquer strategy is that any binary classification algorithm can be used. Besides choosing a decomposition scheme and a classifier for the binary decompositions, one also needs to devise a strategy for combining the binary classifiers and providing a final prediction. The problems of combining binary classifiers have been studied in the computer science literature.7,8 from a theoretical and empirical perspective. However, the literature is inconclusive, and the best method for combining binary classifiers for any particular problem is open.
Standard modern approaches for combining binary classifiers can be stated in terms of what is called output coding. 9 The basic idea behind output coding can be illustrated by the following example: Given three classes, the first classifier may be trained to discriminate classes 1 and 2 from 3, the second classifier is trained to discriminate classes 2 and 3 from 1, and the third classifier is trained to discriminate classes 1 and 3 from 2. Two common examples of output coding are the one-versus-all (OVA) and one-versus-one (OVO), also called all-pairs (AP) approaches. OVA and OVO combinations of binary classifiers have been applied to the analysis of DNA microarray data. 10
In this paper, a generalized OVO combination of binary classifiers has been proposed and applied to multiclass tumour classification. Latent Variable Model (LVM) was chosen as the binary classifier, which has been successfully applied to microarray classification. Results from the proposed OVO method was compared to that obtained using OVA strategy by applying them to two publicly available microarray gene expression datasets Two major categories of feature selection methods have been tested: fold change as well as penalized t-test.
Materials and Methods
Datasets
Two data sets have been used for this comparative study. The first dataset on which the proposed method applied was to the well-known GCM dataset. 4 It consisted of 144 and 54 training and test samples, respectively, representing 14 tumor types. These tumor types included BR (breast adenocarcinoma), PR (prostate adenocarcinoma), LU (lung adenocarcinoma), CO (colorectal adenocarcinoma), LY (lymphoma), BL (bladder transitional cell carcinoma), ML (melanoma), UT (uterine adenocarcinoma), LE (leukemia), RE (renal cell carcinoma), PA (pancreatic adenocarcinoma), OV (ovarian adenocarcinoma), ME (pleural mesothelioma) and CNS (central nervous system). All samples were primary tumors with the exception of eight metastatic tumors in the test set. Expression data was generated using Affymetrix high-density oligonucleotide microarrays containing 16,043 known human genes or expressed sequence tags (EST). The distribution of training and testing samples among the 14 classes is listed in Table 1. The expression intensities for each gene were calculated using Affymetrix GENECHIP analysis software.
GCM dataset: Number of samples per tumor class. a

BR, Breast; PR, Prostate; LU, Lung; CO, Colorectal; LY, Lymphoma; BL, Bladder; ME, Melanoma; UT, Uterus; LE, Leukemia; RE, Renal; PA, Pancreas; OV, Ovary; ML, Mesothelioma; CNS, Brain.
The second dataset (BC) 11 used in this study contained 92 brain cancer expression profiles consisting of 7129 genes using an Affymetrix oligonucleotide array. These samples are grouped into 6 classes: 46 samples of classic medulloblastoma (CMD); 14 of desmoplastic medulloblastoma (DMD); 10 of malignant gliomas (MG); 10 of atypical teratoid/rhabdoid tumors (AR); 4 of normal cerebellum (NC) and 8 of supratentorial primitive neuroectodermal tumors (PN). Ideally we should include all cancer and non-cancer subtypes in the dataset for classification. After performing some preliminary studies, we found that the expression level of the NC group varies significantly from the other cancer related sub-groups. With such large variability and the extreme small size (4 samples) we decided to remove the NC group. Additionally, due the large number of samples in the CMD group (half of all samples) and our concern that it will dominate the classification progress, we decided to remove this group and to focus only on 4 groups with relative equal number of samples. Consequently we used 4 classes of cancer were two of them, when classified solely by morphological characteristics, were in controversy of whether they belong to the same group or not. The distribution of training and testing samples among the 4 subtypes of the BC dataset is listed in Table 2.
BC datasets: Number of samples per subtype. b

DMD, Desmoplastic medulloblastoma; MG, Malignant gliomas; AR, Atypical rhabdoid; PN, Primitive neuroectodermal.
Data preprocessing
Data preprocessing of both the datasets consisted of threshold treatment to the expression intensities with 20 and 1600 as the lower and upper limit, respectively, after which the log2 transformation was applied. Further, genes with the highest transformed intensity being smaller than two times the minimum expression were deleted.
Feature selection (Initial gene pool)
The initial gene pool was build to reduce number of genes or features in a data. Many researches have revealed that when the number of features is large, the performance of the learning methods degrades. Ideally, one would like to select an optimal subset of features that would yield maximum predictive power for a given classification algorithm. In the case of high-dimensional data sets, this can be very computationally demanding; consequently, many statistical and rank based methods are used. In this study, two criteria were used for feature selection. Each of the two criteria was computed for every gene separately.
Fold Change (FC)
At the log2 scale, the fold change is the absolute value of the difference of expression intensity means between two groups. For OVA partitioning of data, this can be expressed as FC = |
Penalized t-statistics
T-statistics is defined as:

where
For genes with very small

In this study, the size of the feature (genes) set used was 50, 75, 100 and 200 genes for the BC dataset and 50, 100 and 200 genes for the GCM dataset.
Binary classification method
Latent variable models are often used in binary classification. It consists of establishing a relationship between the observed binary response,

Further, if a set of exploratory factors,

The link function of the systematic component
Thus,

where ϕ(·) is the standard normal cumulative distribution function. In the case of using expression profiling for disease classification, the matrix

Inferences on the model parameters β, and the point and interval estimates of the quantity of main interest,

Convergence of the sampling process was accessed based on visual inspection of the samples trace plots, and was always reached in less than 200 rounds. Conservatively, a long chain strategy of 20,000 iterations was implemented with the first 5,000 iterations discarded as burn-in. In order to alleviate autocorrelations, one in every 50 samples of the remaining 15,000 draws was maintained for post Gibbs analysis.
Due to the fact that the number of genes is much greater than the number of samples; a dimension reduction technique called singular value decomposition (SVD) is performed before fitting the regression model. For the BC dataset, five replications were done by randomly selecting the training samples and testing samples from each subtype by maintaining the same number of samples in both training and testing samples as mentioned in Table 1. For the GCM dataset, exact split done by Ramaswamy et al 4 was done.
Output coding strategies implemented for multiple classifications
Once the discriminative genes (features) are selected based on fold change and penalized t-test, samples that contain only those selected (top ranked) genes were used for training as well as for testing. The two output coding strategies implemented in this study using LVM (binary classifier) are described below.
One-versus-all method
The one-versus-all approach
4
divides the classes into two groups each time, with one group consisting of a single class and the other group consisting of samples in all the other classes. In other words, given
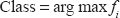
where
For example, in this study, if we take DMD as one group, the other group will contain the remaining samples from MG, AR and PN. The DMD samples will be given the binary status 1 and all other tumor samples except DMD will have 0 as binary status. The predictive ability of LVM was tested using a cross-validation procedure using the test samples (Tables 1 and 2), in which the binary response for one test sample was treated as unknown and the regression coefficients were calculated using the training samples of the 2 tumor types. The estimated coefficients were then used to predict the status of the unknown test sample. This process is repeated for each test sample of all tumor subtypes.
One-versus-One method
This approach involves implementing LVM for each pair of tumor classes. This ensures that each of these groups is compared with each of the remaining groups one by one, and the corresponding selected genes can represent the most significant differences. The usual way to perform this is, given
Proposed One Versus One (OVO) output-coding strategy.
Let A, B, C & D represents four tumor types
The predictive ability of LVM is tested using cross validation procedure, in which the binary response for one test sample is treated as unknown and regression coefficients are calculated using the training dataset (only 2 classes at a time).
For a particular test sample (for ex. A), cross validation procedure is implemented for each pair wise combination to estimate P(Y

Represents the binary status of the training data while implementing LVM.
Mean and median of each column (for ex. column 1 contains the probability that the test sample is predicted to be A compared to all other tumor classes (pair wise comparison)) is estimated and the test sample will be assigned to that particular tumor class whose estimated mean or median is highest. For example, if column 1 has the highest mean compared to other 3 columns then the test sample will be classified as tumor class A.
Steps 1, 2 and 3 are repeated for each test sample for all tumor classes.
Results
Prediction accuracy determination of the optimum number of genes (features) to be used by the classification algorithm is usually a difficult task that depends on several factors including the classifier rules and the complexity of the data set. Finding the optimal number of genes is generally very difficult. Many practical solutions are based on experience or some heuristics. 14 For binary regression algorithms, previous studies.15,16 showed a feature set of one to two hundred top genes was adequate for a simple two-category problem. In this study, the size of the feature set was 50, 75, 100 and 200 for the BC dataset and 50, 100 and 200 for the GCM dataset.
BC dataset
The prediction accuracies when OVA and OVO output-coding strategies were implemented for the 5 replications using fold change and penalized t-test for feature selection are summarized in Tables 3 and 4 respectively. The highest average prediction accuracy of the 5 replications obtained for fold change for OVO was 93.33% for 75 genes, where as, for OVA the highest average was 83.33% for 75 genes. For penalized t-test, the highest average for OVO was 91.66% for 50 genes while, for OVA, the highest prediction accuracy was 81.67% for 200 genes. This result shows that OVO strategy could give a higher prediction accuracy compared to OVA. Also features (genes) selected by fold change gained highest accuracies for both OVO and OVA. There was an improvement of around 12% by using OVO method compared to OVA method for fold change and 15% improvement for penalized t-test for the BC dataset. Leung et al 16 got 17% improvement by using OVO over O VA using t-test for feature selection and k-means clustering for classification.
Prediction accuracies for the BC dataset using fold change as the feature selection method.

Prediction accuracies for the BC dataset using penalized t-statistics as the feature selection method.

GCM dataset
The prediction accuracy of the 54 validation (test) samples, using fold change and penalized t-test, is summarized in Tables 5 and 6 respectively. The highest prediction accuracy for OVO with fold change feature selection was 75.92% for 50 genes where as for the same number of genes and fold change, OVA performed higher (79.6%). For 200 genes, the fold change feature selection method performed equally for both OVO and OVA (70.4%). The highest prediction accuracy for OVO with penalized t-test was 74% for 50 genes where as an accuracy of 79.6% was obtained for OVO method for 100 genes when the second highest mean was also considered for each sample (data not shown). For 200 genes selected based on penalized t-test, the OVO had a higher accuracy of 3% compared to OVA, which is not that significant. But for 50 genes and penalized t-test showed higher performance in OVA compared to OVO.
Prediction accuracies for the GCM dataset using fold change as the feature selection method.

Prediction accuracies for the GCM dataset using penalized t-statistics as the feature selection method.

Several works have been done using this GCM dataset using various feature selection methods and classification methods on OVA set up. Using a recursive feature selection procedure and support vector machine (SVM) classification algorithm, Ramaswamy et al 4 obtained their best result with 42 tumors correctly predicted among the 54 test samples, corresponding to an accuracy of 78%. Using a feature selection algorithm based on overlaps of gene expression values between different classes in conjunction with the Covering Classification Algorithm (CCA), a modification of the k-NN method, Bagirov et al 17 achieved prediction accuracy of around 80%. Based on the concept of gene interaction, Antonov et al 18 proposed a Maximal Margin Linear Programming (MAMA) procedure that combines linear programming and SVM and they got around 85.2% classification accuracy on an OVA set up. Combining all these previous research and the results obtained from this study, it may be concluded that for GCM dataset, OVA performs almost equally as that of OVO.
Discussions
Classification problems aim at building an efficient, effective model for predicting class membership from the data. The learning process is done on the training data, which consists of data points chosen from the input data space and their class label. A model built from the training data is expected to produce the correct label on the training data and also to predict correctly the identity of an unknown class. In cases where there is only two classes, a classification problem is said to be a binary, while many real-world problems are multi-class problems. Majority of the solutions for multi-class problems is done by decomposing the multiple classes into binary ones. One-versus-the rest methods, pair wise comparison, error-correcting output-coding (ECOC) 20 are examples. The main criticism against OVR is because of the inconsistent class assignment as well as solving potentially asymmetric problems using a symmetric approach..14,21 OVO also can be criticized for solving asymmetric problems symmetrically, but one advantage of using this method is that each classifier is easy to train since it is purely a binary problem even though the number of comparisons will be higher than OVR.
The usual way of implementing OVO is based on vote, which class label gets the largest number of votes, that test sample will be assigned that class label. While the OVO implemented in this study uses summary of the posterior distribution (mean and median) of the classification probability to determine the fate of the test sample.
Latent Variable models (LVMs) postulate that the observed discrete (binary data) is only an indicator of an underlying non-observed random variable that follows a certain distribution (often normal distribution). The discrete responses are observed when the latent variable exceeds a certain threshold(s). For example for the binary situation, the binary response is 1 (case) when the latent variable exceeds threshold, T, (i.e. T > 0) otherwise it is zero (control). The main advantage of using LVM along with the proposed OVO in the context of a Bayesian implementation via Gibbs sampler is because its ability of providing the full posterior distribution of the classification probability..13,15,22 Other binary classifiers like SVM will output only the class label (+, or –) of the test sample. However, knowing the class label or the predictive value a lot of times is not good enough to evaluate a classification. This makes LVM a good classifier that suits well with the proposed OVO strategy.
It has been mentioned that there is probably no multiclass method using binary decomposition approach that outperforms every thing else and that for practical purposes the choice of the method has to be made depending on the constraints, such as the desired level of accuracy, the time available for development and training, and the nature of the classification problem.
6
The same conclusion has been made in the current study since different gene expression data sets showed differences in the prediction accuracies. In OVO decomposition strategy, we need to do 2x(
Number of misclassified test samples of Breast (BR) and their predicted tumour subtype according to OVO for both fold change and penalized t-statistics.

In parenthesis is the assigned tumour types for the misclassified BR test samples.
Conclusions
The output-coding scheme from machine learning has been successfully applied to multi-class microarray classification in this paper. It has been shown that a good coding matrix can lead to high accuracy of multi-class microarray classification. Better coding strategies are required to further improve the performance of the output coding scheme. This study demonstrated that the choice of feature selection statistics, comparison method between groups and classification algorithms could all affect the interpretation of final results of microarray data. More emphasis is given on the comparison methods and it could be concluded that the selection of OVO or OVA depends upon the data structure and the type of microarray experiment. Based on the BC dataset, we can assume that when dealing with multi-class cancer type datasets, OVO comparison method can give better performance accuracy than the commonly used OVA. But we could not conclude the same inference with the GCM dataset. The main reason could be: large number of tumor types is considered at the same time or may be because of the heterogeneity within and among the cancer subtypes or chances of potential mislabeling of tumor subtypes in the training data.
Disclosures
This manuscript has been read and approved by all authors. This paper is unique and is not under consideration by any other publication and has not been published elsewhere. The authors report no conflicts of interest.